CMN Weekly (11 July 2025) - Your Weekly CRISPR...
CMN Weekly
Jul. 11, 2025


Leber congenital amaurosis (LCA) is a group of rare and incurable inherited retinal diseases that manifest at birth or in the first few months of life and progress over time, leading to blindness by the 3rd or 4th decade of life.
LCA is an autosomal recessive disease arising from loss-of-function mutations in any one of at least 27 genes. Mutations in many of these genes result in dysfunctional rods and cones, which are the light-gathering cells of the retina, and mutations in the RPE65 and CEP290 genes are among the most prevalent. Disease severity depends on the exact mutations present.
RPE65 is a critical isomerase in the classical visual cycle in vertebrates that regenerates the active visual chromophore 11-cis-retinal. The function of CEP290 is not entirely understood but it appears to play a role in the centrosomes and cilia, which are important for cell division and perception of sensory input, respectively.
LCA is estimated to occur in 1-2 out of 100,000 live births worldwide, and until the approval of the gene replacement therapy Luxturna (first in the US in 2017, and in the EU in 2018), treatment was limited entirely to symptom control and supportive care. Although rare, LCA is the most common cause of childhood blindness.
Luxturna, developed by Spark Therapeutics (since acquired by Roche), is based on sub-retinal injection of a functional RPE65 cDNA delivered via an adeno-associated virus (AAV) and represents the first dedicated therapy for LCA. It improves vision in some individuals but is not curative and concerns exist about its long-term efficacy, with reports of continual retinal degeneration after 1-3 years. Furthermore, its use is restricted to a subset of children and adults that have mutations in both RPE65 alleles and who have a certain threshold of retinal function at the time of treatment.
The world’s first ever in vivo CRISPR therapy is currently being evaluated in the BRILLIANCE clinical trial for LCA10, for which no treatments exist as of yet.
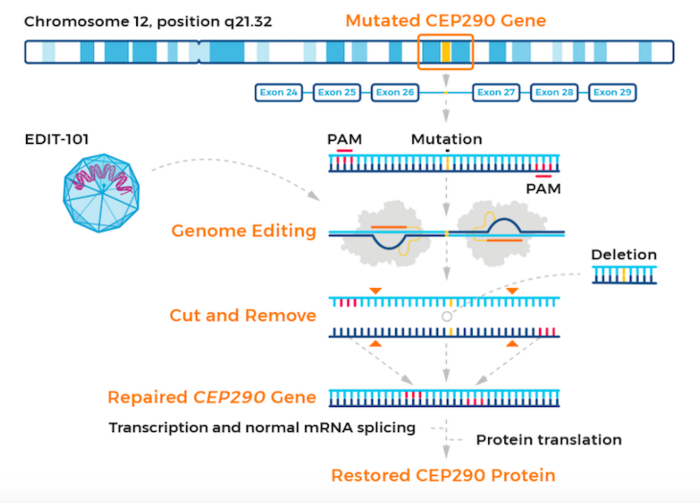
The therapy, known as EDIT-101, is developed by Editas Medicine, US. EDIT-101 is based on CRISPR-Cas correction of mutations in the CEP290 gene. Mutations in this gene are responsible for LCA10, which accounts for 20-30 % of all LCA cases.
EDIT-101 is designed to restore normal CEP290 protein expression, photoreceptor function and vision. Specifically, EDIT-101 is a CRISR-Cas9-based gene-editing candidate designed to remove the IVS26 mutation from the CEP290 gene. EDIT-101 comprises an adeno-associated virus serotype 5 (AAV5) vector containing a Cas9-encoding gene and two guide RNAs designed to remove IVS26.
EDIT-101 is adminstered to patients via a sub-retinal injection to reach and deliver the gene-editing machinery directly to photoreceptor cells. While CEP290 mutations disable light-detecting cells in the retina, the retinal cells themselves are still present and viable in LCA10, raising hopes that EDIT-101 can improve vision in this patient group.
The trial is expected to enrol a total of18 paediatric (3-17 years) and adult individuals with LCA10 who have a certain baseline level of visual function. The study is open-label and will be run in multiple centers over five cohorts that will each receive one of three doses that will be administerd via subretinal injection.
Dosing in the low-dose cohort was completed in November 2020, and Editas reported in February 2021 that it had initiated dosing in the mid-dose cohort.
In September 2021, Editas Medicine shared positive initial clinical data from adults dosed wth EDIT-101 in the BRILLIANCE trial. At that time, Editas revealed that adult patients treated with EDIT-101 had not experienced serious adverse events or dose-limiting toxicities, and modest signs of visual improvements were reported by patients in the mid-dose cohort.
In April 2022, Editas Medicine revealed in a press release that it had initiated dosing in the paediatric mid-dose cohort in the BRILLIANCE trial following endorsement of an Independent Data Monitoring Committee (IDMC). This endorsement is based on an analysis of clinical safety data from adults in the low- and mid-dose cohorts who have already received EDIT-101. According to the same press release, the company remains on track to complete testing of the paediatric mid-dose in the first half of 2022 and it also expects to initiate testing of the paediatric high-dose this year.
Further clinical updates including safety and efficacy assessments for all adults dosed in the BRILLIANCE trial, who have had at least six months of follow-up evaluations, are expected in the second half of 2022.
Krzysztof Palczewski’s group at the University of California Irvine (UCI) is exploring base editing as a new treatment modality for the LCA2 subtype, which is caused by mutations in the RPE65 gene and accounts for approximately 16% of all LCA cases.
In work published in Nature Biomedical Engineering in late 2020, the group demonstrated for the first time that base editing can correct an LCA2-causing nonsense mutation in vitro and in vivo, with restoration of visual function in a mouse model for retinal disease. We interviewed lead author of that study Susie Suh, an MD PhD student from the Palczewski lab shortly after the work was published, who told us more about this proof-of-concept study.
In April 2022, researchers from the same group published new findings in Nature Communications, demonstrating that in vivo correction of an RPE65 mutation by an adenine base editor (ABE) could prolong the survival of cones in an LCA2 mouse model, which is important given the progressive deterioration of retinal photoreceptors observed in patients years after receiving the currently available gene therapy.
Although efforts to develop base-editing and prime-editing therapies for LCA are still in their infancy and pre-clinical testing is necessary in non-human primates before any new therapeutic candidate can enter the clinic, the studies undertaken to date provide hope that these gene-editing modalities could be a feasible option for the treatment of LCA in the future.
As the CRISPR field advances, it is likely that base-editing and prime-editing therapeutic strategies for LCA will move towards clinical development, and we will continue to bring updates as they come.
In the meantime, you can get a complete overview of the current gene-editing clinical trials in our CRISPR Medicine News Clinical Trials Database.
This roundup was originally published on 24th November 2021. It was updated on 8th June 2022 to reflect further developments in the CRISPR medicine field for Leber congenital amaurosis.
To get more of the CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.
ArticleClinical News UpdatesHereditary BlindnessLeber Congenital AmaurosisEditas Medicine, Inc.