IND Enabling
Phase I
Phase II
Phase III

ICD-10 Disease Code: D84.1
Defects in the complement system
ICD-10 Disease Group: D84 Other immunodeficiencies
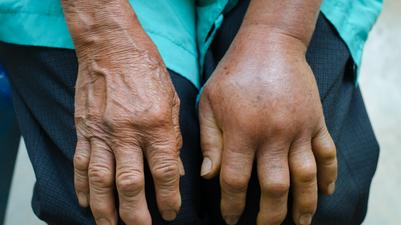
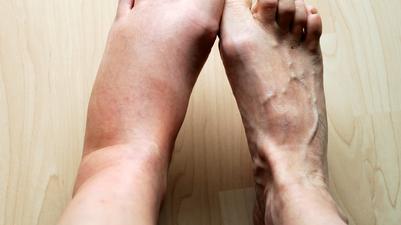
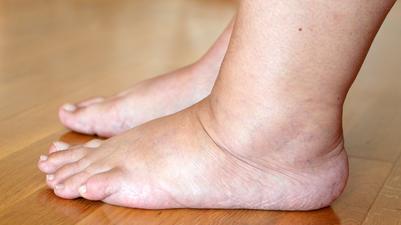
General description:Hereditary angioedema (HAE) is a rare genetic disease that is characterised by severe, recurring and unpredictable inflammatory attacks with swelling in organs and tissues throughout the body.
The disease arises from lack of a protein called complement component C1 esterase inhibitor. This protein plays an important role in controlling inflammation in healthy individuals.
HAE attacks may be painful, debilitating and life-threatening.
Current treatments include a number of therapies that aim to replace the missing protein to correct the deficiency, or drugs that inhibit another protein called plasma kallikrein. Inhibiting plasma kallikrein prevents the production of an inflammatory molecule called bradykinin, which helps to reduce pain and swelling as well as prevent disease attacks.
These treatments are life-long, and may require once- or twice-weekly injections, or daily oral administration to ensure constant disease control. Despite chronic administration, breakthrough attacks still occur, and the disease remains incurable.
Mutations:HEA type I is an autosomal dominant disease that arises from a deficiency in a protein known as complement component C1 esterase inhibitor, which is important for controlling inflammation. Specifically, mutations in the SERPING1 gene cause the two main disease types HEA type I and HEA type II. The SERPING1 gene is located on the long arm of human chromosome 11 (11q12-q13.1).
HEA type I accounts for about 85 % of all cases. HEA type II is a more rare form of the disease, occurring in about 15 % of cases, and arising from abnormal C1 esterase proteins that do not function properly.
Disease frequency:HEA is estimated to affect between 1 in 50,000 to 1 in 150,000 individuals worldwide.
HEA affects males and females in equal numbers.
Symptoms:The symptoms seen in HEA develop result from a deficiency or subfunctional complement component C1 esterase inhibitor, whose physiological role is to maintain the normal flow of fluids through the blood capillaries. HEA is characterised by recurrent episodes of the accumulation of fluids outside of the blood vessels, and occasioanlly within internal organs. This blocks the normal flow of blood or lymphatic fluid and causes rapid swelling (oedema) of tissues in the hands, feet, limbs, face, intestinal tract, or airway. Swelling usually occurs without itching, but may be painful. Swelling in the gastrointestinal tract leads to cramping, while swelling of the airways may lead to obstruction, a potentially very serious complication. Symptoms can occur at any age, although they typically begin in early childhood, and disease severity varies greatly among affected individuals.
Treatment:Current treatments include a number of approved replacement therapies using preparations of C1 esterase inhibitor to correct the deficiency, or drugs that inhibit plasma kallikrein, which in turn halts the production of the inflammatory mediator bradykinin to reduce pain and swelling as well as prevent HAE attacks. The disease is incurable.
Sources: