CMN Weekly (26 July 2024) - Your Weekly CRISPR...
CMN Weekly
Jul. 26, 2024


Base-editing systems have been hailed as safer alternatives to traditional CRISPR-Cas9 gene editing, primarily because they can induce point mutations without creating double-stranded breaks.
Despite the clear potential of these systems in correcting single nucleotide polymorphisms (SNPs) - which cause a majority of human genetic diseases - a major stumbling block in the therapeutic application of base editors (BEs) is their off-target activity.
In a study published last month in Nature Chemical Biology, a team of researchers from Associate Professor Rahul Kohli’s lab at the University of Pennsylvania reveal the first controllable base-editing system.
Kiara Berríos, a senior Ph.D. candidate in the Kohli lab and the first author of the study, had no intention of doing a PhD in gene editing, but her interest in DNA-modifying enzymes led her to develop this key breakthrough in the base-editing field.
»This is special because it takes you to the next level of control – manipulating the time that base editors are active,« Berríos says.
At the time the first study about base editors (BE) was published by David Liu’s lab at Harvard in 2016, Kohli’s lab had actually been studying the enzymes used by BE systems for years - but in a completely different context. The group of proteins that facilitate transition mutations in BE systems is the activation-induced cytidine deaminase (AID)/apolipoprotein B editing complex (APOBEC) family, and as an infectious disease doctor, Professor Kohli had been researching the role of AID in the immune response.
The problem with currently available BE systems is that the DNA deaminase enzymes used, like AID/APOBEC, are unregulated; once administered, BEs will induce transition mutations across the genome and transcriptome, independent of their sgRNA.
»When I started the project, the studies about base editing being dangerous and the off-target effects weren’t published yet. But we knew that would be the case, because of what happens when these enzymes are misregulated in vivo – when they’re overexpressed or misregulated they cause cancer, so we knew that if you overexpress base editors you would get that kind of activity as well,« Berríos explains.
The Kohli lab members wanted to use their knowledge of these enzymes to develop safer base editors by creating layers of control, teaming up with Assistant Professor Junwei Shi’s lab, also at UPenn. The idea was to split the deaminase enzymes and find a system that could reconstitute the split protein and then induce their activity – similar to the split-Cas9 system developed by Feng Zhang’s lab in 2015.
For many scientific breakthroughs, the critical, nail-biting moment comes towards the end of months or years of experiments. For Berríos, it was two of the earliest assays in her study. First, Berríos needed to prove that DNA deaminases used for base editing would tolerate insertion of a reporter molecule at a particular site. An initial assay in bacteria demonstrated that AID/APOBEC with green fluorescent protein (GFP) inserted in the α2–β3 loop retains the same amount of activity as the original enzyme.
The next critical step was to split the protein and see if the two halves were inactive and if their activity could be reconstituted when brought back together, for which the team used a GFP that self-reassembles in solution, splitting the enzyme at the GFP insertion site.
»When we saw that the insertion was possible and we still got activity [of the enzyme], we were sure it was going to work. After we knew the halves were inactive, then the big moment of truth was actually when we purified the protein and I saw that the GFP was together and it was fluorescent – that was amazing,« Berríos says.
Once they’d proven the feasibility of splitting and reconstituting the original AID enzyme, Berríos and her colleagues wanted to find out if other deaminase enzymes could be manipulated in the same way. Fortunately, the AID/APOBEC family of DNA deaminases share very similar structures, so their experiments on the commonly used APOBEC3A (A3A) enzyme were similarly successful.
»We thought we could do it because they all share the same fold. The only difference is that the loops are a bit longer or shorter depending on what enzyme we’re talking about. So because they all share that fold, we thought if we could split one of them at this site, we should be able to split all of them,« Berríos elaborates.
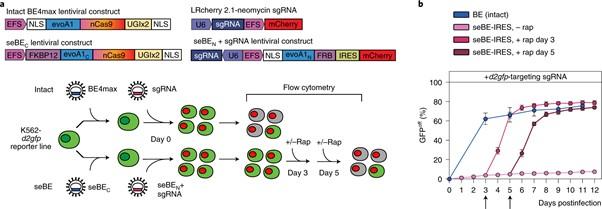
Berríos and the team then needed to find a drug-inducible system – a molecular ‘on switch’ – that could be used to control the split-engineered base editors (seBEs). To do so, they employed the FK506-binding protein 12 (FKBP12) and FKBP rapamycin-binding domain (FRB) complex, a common protein dimerisation system that may be induced using the small molecule rapamycin (See Figure 1 for a graphical overview of the approach).
»Originally, we had the split GFP, which was what was bringing the halves [of the enzyme] together. But we couldn’t control that, so we wanted to find another dimerisation system that we could induce with a small molecule. That’s when we switched to FRB and FKBP12 – when we add rapamycin they come together,« Berríos explains.
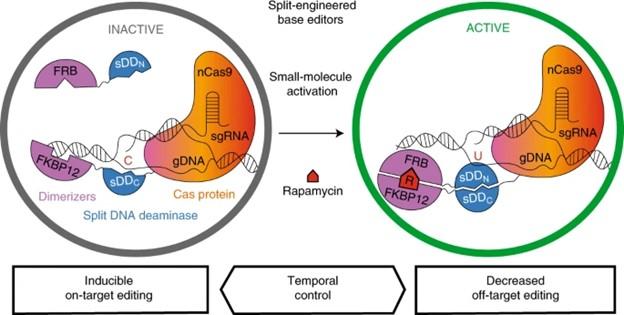
Berríos and her colleagues now had a controllable, inducible system of BEs, but they still needed to prove that the seBE-T2A construct induced with rapamycin had similar levels of efficiency to the original BE systems. After confirming that seBEs could robustly inactivate destabilised GFP (d2gfp) in a HEK293T reporter cell line, they tested the system across a broader range of genomic targets and assessed the off-target activity.
»We used [the T2A construct] on nine different targets sites. It retained the activity and was basically the same as the intact editing system, only slightly lower in efficiency, but that could also be due to a slight difference in timing, because the intact base editors start editing immediately, while the seBEs are only activated after 24 hours,« Berríos explains.
The team also used an assay developed by David Liu’s lab to assess potential off-target effects of seBEs and compare these to intact BE systems. They co-expressed the intact BE4max and seBE-T2A constructs with catalytically-dead SaCas9 (dSaCas9) – which opens the DNA double helix to form the R-loop but does not induce double-stranded breaks – in HEK293T cells.
»That assay was great, because we could see the intact base editors had around 10% off-target activity, but the seBEs in, both in the absence and presence of rapamycin, had very low levels,« Berríos comments.
An unexpected result was some low-level activity of the seBE-T2A construct in the absence of the rapamycin, however Berríos and her colleagues identified the cause of the problem and addressed the issue by using a different construct, which they are also employing in continuing work.
»The T2A construct is a ribosome-skipping peptide. If the ribosome doesn’t skip it, then it keeps going and makes the whole protein, so that’s why we were seeing the activity in the absence of rapamycin. But then we switched to a different construct, seBE-IRES, where the sections were separated by a stop codon. That reduced the activity significantly, to about 1%, but I think we can get it down to zero,« she remarks.
One of the final hurdles was to assess whether seBEs could really be incorporated into living cells, lying dormant for extended periods of time before being activated when desired. After lentiviral transduction of the seBE-IRES fragments into the K562 leukaemia reporter cell line, minimal editing was observed over a period of 12 days. When rapamycin was added to cells at varying intervals, however, the seBE-IRES construct was able to perform editing of d2gfp at levels approaching those seen by the intact BE4max system (Figure 2).
Berríos says the strong temporal control over seBEs evidenced by these experiments will provide ideal conditions not only for therapeutic use, but also for disease modelling. By controlling the timepoint at which editing occurs, researchers can target certain developmental stages, or critical steps in cancer or disease pathogenesis.
Berríos and her colleagues are still hard at work in the lab, trying to make the seBE system as efficient and safe as possible and demonstrating its use in vivo.
»We’re still optimising how much rapamycin to add so that our system is active just the right amount of time to achieve optimal on-target efficiency and limited off-targets. The hope is that we can find a condition with optimal on-target activity and no off-targets. Then we’re planning to continue the work in mice,« she says.
The team also hope that future work will build on their study, adding more layers of control to base-editing systems so that they can be spatially as well as temporally controlled. And the significance of the seBE system doesn’t end in the clinic – as Berríos explains, split-engineered deaminases will also be useful in other areas of research.
»We have a lot of problems studying DNA deaminases because they’re toxic to cells, but if you use a split system, you can study them without them causing toxicity. You can also study cancer and somatic hypermutation, because you have a way to control and induce these processes.«
The future is looking bright for Berríos. Just months away from completing her Ph.D, she has already been accepted as a postdoctoral research fellow in Professor David Liu’s lab at Harvard. Despite joining the Kohli lab with an intention of studying the biochemistry of DNA deaminases and their role in the immune system, she couldn’t be happier with where her career path is leading her, and it’s a lesson in serendipity.
»When I first started working in this lab, I was really interested in how proteins interact with DNA – to understand that process from a biochemical perspective, and how proteins can change DNA. So it’s super interesting to be able to harness that power for base editing. I wasn’t expecting it, but I guess I was interested in base editing all along without really knowing it,« Berríos concludes.
Link to the original paper published in Nature Chemical Biology: Controllable genome editing with split-engineered base editors.
Rebecca Roberts is a molecular biologist and science writer/communicator based in Queensland, Australia.