CMN Weekly (26 July 2024) - Your Weekly CRISPR...
CMN Weekly
Jul. 26, 2024


Despite being the second most common form of congenital muscular dystrophies, there are currently no effective treatments available for collagen VI-related dystrophies (COL6-RDs), a fact that Cecilia Jiménez-Mallebrera and her research team hope to change using CRISPR technology.
Jiménez-Mallebrera has been studying neuromuscular diseases for almost her entire career, specialising in ultra-rare congenital muscular dystrophies in paediatric patients. Currently the director of the Laboratory of Translational Research in Children’s Neuromuscular Diseases at IRSJD, her work spans the pathophysiology and diagnosis of these conditions through to clinical trials.
Her recent paper, published in the International Journal of Molecular Sciences, demonstrates the use of CRISPR-Cas9 gene editing to alleviate the pathogenic effects of a dominant negative mutation in the COL6A1 gene.
As postdoctoral researcher and first author of the paper Arístides López-Márquez explains, this work is a key development towards the treatment of diseases with this specific mutation, but it also has significant implications for other congenital muscular dystrophies:
»For this study we worked on collagen VI-related disorders, but our aim is that in future this strategy could be applied for other congenital muscular dystrophies. All the results that we have obtained and the optimisation of the protocols are going to be really useful for similar mutations involved in other diseases.«
COL6-RDs are caused by mutations in the COL6A1, COL6A2 and COL6A3 genes that encode the alpha chains of collagen VI, a key component of the extracellular matrix (ECM). Pathogenic mutations in these genes negatively affect the assembly of the collagen VI microfibrillar matrix, resulting in weakness and wasting of the muscles.
COL6-RDs range from mild, as seen in Bethlem myopathy, to the much more severe Ullrich’s congenital muscular dystrophy; with no effective treatments, patients experience progressive and often debilitating disease. Jiménez-Mallebrera has been working with many of these patients for a number of years, allowing her to choose a viable target for genetic therapy.
»We have a cohort of patients that are very well characterised from the genetic point of view, and also from the clinical point of view. For this study, we focused on a very specific mutation which is very common in COL6-RDs. It’s a glycine substitution in the COL6A1 gene, which is very important for the formation of the mature molecule. Because it is a common mutation we started working on this mutation first,« Jiménez-Mallebrera explains.
The prevalent pathogenic mutation in COL6A1 is a single nucleotide substitution, where a glycine is replaced by an arginine in the N terminal of the triple helix domain (c.877G>A; pGly293Arg). This affects the folding of the resulting protein, hindering the association of tetramers to create the necessary collagen VI microfibrils. The mutation is dominant-negative, meaning that only one copy of the mutation is required to cause disease and the mutant allele interferes with the production of protein from the wild-type allele.
With patient outcomes foremost in her mind, Jiménez-Mallebrera wanted the results of the study to be as clinically relevant as possible. To achieve this, she leveraged fibroblasts from the well-characterised patient group she has been working with for years, obtaining cells from four different patients carrying the same COL6A1 mutation and with varying levels of collagen VI and disease severity.
The original goal of the study was to correct the pathogenic mutation in COL6A1 via homology-directed repair (HDR) using a donor DNA template. The team assessed two different guides targeting opposite strands at exon 10 of the COL6A1 gene to determine which would provide the best editing outcome, and included a silent mutation in the DNA donor template to avoid re-cutting at the same site after editing.
Unfortunately, Jiménez-Mallebrera and the team quickly realised that this strategy was not effective; after sequencing to confirm the experimental results, the team saw that HDR had occurred at frequencies of less than 1%. Yet this failure quickly revealed itself to be a blessing in disguise; more than 40% of the sequencing reads bore frameshift mutations that resulted in silencing of the mutant allele, with no effect on the wild-type allele.
Following editing experiments, the team performed digital droplet PCR (ddPCR) with allele-specific probes to determine the expression level of each allele. While expression of the mutant allele was significantly reduced – by more than 80% - the wild-type was expressed at normal levels.
Theoretically, silencing of the mutant allele could be a therapeutic alternative to correction because it would no longer interfere with the wild-type allele. The next step was to determine whether silencing of the mutant allele was enough to recover the function of the fibroblast cells in forming the collagen VI matrix.
»We want to silence the mutant allele because this is a dominant-negative mutation. The mutated collagen VI chains can associate with the wild-type chains, but once they are in the ECM they impair the formation of the collagen VI network. Most of the mutations in collagen VI are dominant, so silencing the mutant allele permanently at the genomic level is efficient. By doing so you can replace the mutated chains in the extracellular matrix, increase the proportion of the wild-type chains, and thereby restore the structure of the cellular matrix,« Jiménez-Mallebrera says.
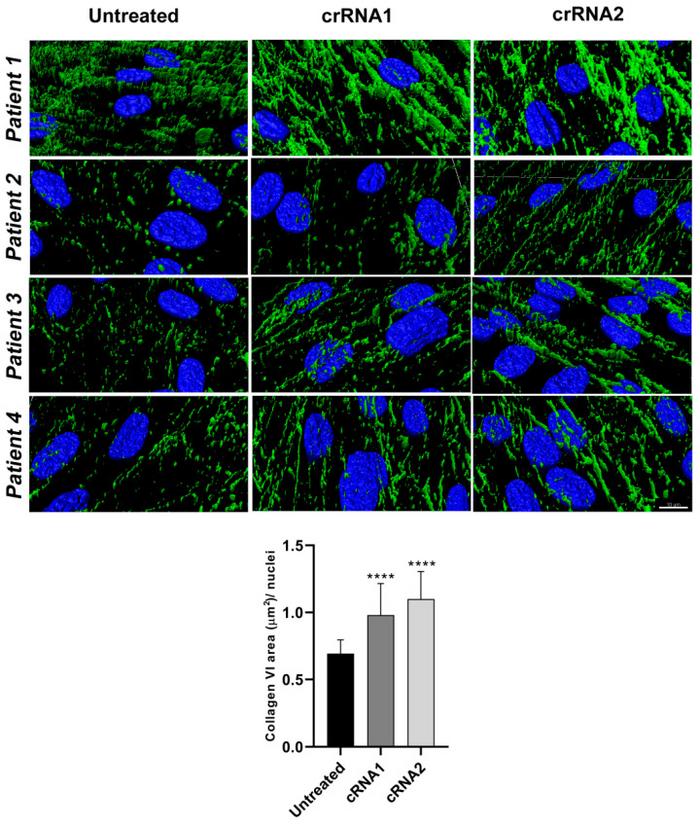
Immunofluorescence assays and confocal super-resolution microscopy helped the team examine the effects of COL6A1 mutant silencing on the collage VI microfibrillar ECM. Overall, they observed significant recovery of the collagen VI signal in the edited patient cells, with thick and connected collagen VI fibers similar to those seen in control cells (Figure 1).
An obvious obstacle for this type of therapy is delivering CRISPR-Cas9 reagents to fibroblasts in vivo; successful editing of somatic tissues such as muscle remains a significant challenge in the broader field. ButJiménez-Mallebrera and her team are determined to find a viable strategy to deliver this potentially life-changing therapy. They hope recent progress in several areas of CRISPR delivery vehicles, including muscle-trophic adeno-associated virus (AAV) vectors and tissue-specific nanoparticles, might be the answer.
»Now we have verified that CRISPR-Cas9 is useful for this pathology, and we can silence the mutant allele, we want to move to the next step, which in vivo experiments. We want to try both strategies - nanoparticles and viral vectors,« López-Márquez elaborates.
In addition to their work on gene silencing, the team are also investigating other techniques, such as base and prime editing, to correct the mutation. The team have also developed techniques that have allowed them to monitor the efficiency of the editing and its effect on cellular phenotype – something that is critical for these types of studies and was previously a challenge.
»We've developed advanced microscopy techniques to look at the structure of the collagen six fibrils. We also published a deep learning algorithm that can classify the images from the studies of fibroblasts in either patients or controls. So we have a variety of tools to monitor the efficiency of the therapy, which is very important,« Jiménez-Mallebrera remarks.
As Jiménez-Mallebrera concludes, her dedicated research team are trying to cover all possible bases to improve the lives of the patients she has worked with for so many years:
»We're a very translational group, we are very aware that we need to provide a complete solution, not just high editing efficiency at the cell level or even in an animal model. We’re very keen on establishing a strategy that takes everything into account in terms of delivery and safety. It's very important to us that we investigated all these different aspects, so that we can really translate this into a viable gene therapy in the short to medium term.«
Link to the original article in the International Journal of Molecular Sciences:
Rebecca Roberts, Ph.D. is a molecular biologist and science writer/communicator based in Queensland, Australia.