FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...
Loss-of-function mutations in the DYSF gene cause autosomal recessive muscular dystrophies including the rare subtype limb-girdle muscular dystrophy 2B (LGMD2B).
Specifically, LGMD2B results from a common frameshift mutation in DYSF exon 44 that leads to complete loss of the plasma membrane repair protein dysferlin. Dysferlin is involved in the repair of muscle fiber membranes, and its absence results in progressive skeletal muscle degeneration and muscle wasting. Since no specific therapies exist for LGMD2B, treatment plans are usually focused on managing symptoms to improve the quality of life for those affected.
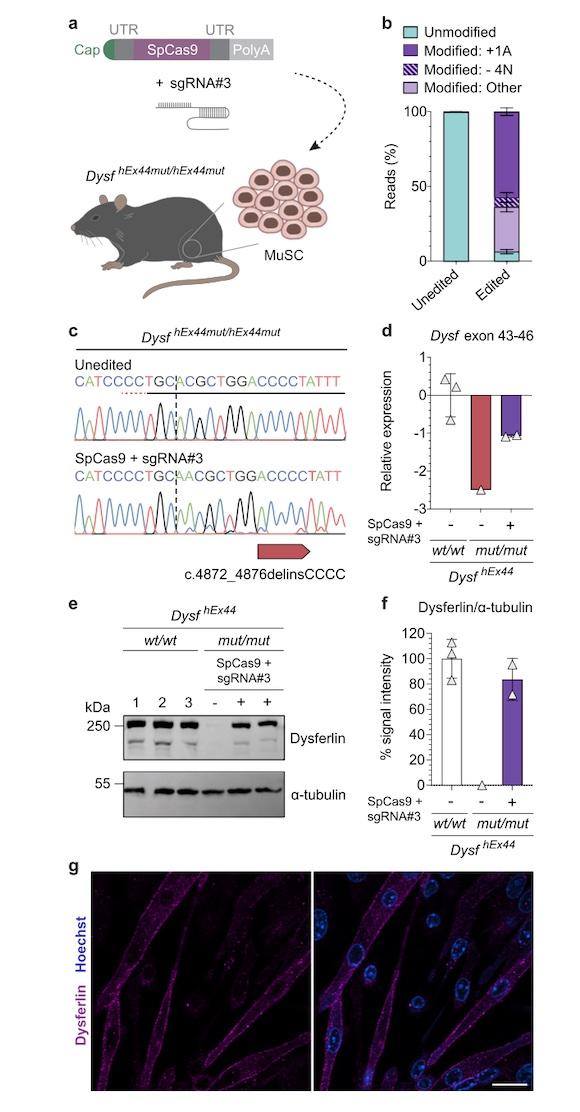
In a recent article in Nature Communications, researchers in Germany report for the first time a CRISPR-based gene correction strategy to restore dysferlin function. The team, led by Simone Spuler MD, Professor of Myology and Group Leader at Max Delbrück Center in Berlin, developed a targeted CRISPR-Cas9 approach that specifically corrects an exon 44 founder frameshift mutation using sgCas9 mRNA and a mutation-specific sgRNA. This method achieved over 60% correction efficiency and restored full-length, functional dysferlin protein in patient and humanised mice primary muscle stem cells.
In an attempt to restore dysferlin function, the team designed a sgRNA targeting a specific founder frameshift mutation in DYSF exon 44 (c.4872_4876delinsCCCC). SpCas9 was delivered as mRNA, and relying on non-homologous end joining (NHEJ) to repair the DNA after Cas9 cleavage, the strategy was designed to result in insertion of a single adenine nucleotide to restore the DYSF reading frame.
The team tested their approach in multiple formats, including: human induced pluripotent stem cells (hiPSCs) derived from two homozygous patients, primary muscle stem cells (MuSC) from patients, and a newly developed mouse model carrying the mutant human DYSF exon 44.
Remarkably, NGS analysis of post-edited primary patient MuSC revealed more than 90% on-target editing, with the desired A insertion in more than 60% cells. This level of correction was concomitant with restoration of full-length dysferlin production, which showed proper localisation within the cells and maintained its normal functional properties.
To validate their findings in vivo, the team developed a new mouse model for LGMD2B that harbours the human DYSF exon 44, with and without the target mutation in the endogenous Dysf locus.
Experiments to characterise the new model confirmed that both humanised wild-type and mutated exon 44 were normally spliced in mouse muscle, and that mice homozygous for the humanised wild-type allele showed normal dysferlin mRNA and protein expression and localisation patterns and no pathological phenotypes.
On the other hand, mice homozygous for the humanised mutated exon 44 displayed phenotypes that aligned with muscular dystrophy, very reduced dysferlin mRNA expression levels and no dysferlin protein expression.
When edited patient MuSC were transplanted back into the newly developed mouse model, the team demonstrated that they successfully generated new muscle tissue, restored normal dysferlin expression and localisation, and could repopulate the MuSC population.
This study represents a crucial milestone as the first in vivo proof-of-concept demonstrating that CRISPR-edited muscle stem cells could potentially be used for autologous cell replacement therapy in muscular dystrophy. The research also introduced a valuable new mouse model for LGMD2B, which will be instrumental for future studies investigating mRNA-based gene-editing or other therapeutic approaches for this incurable form of dystrophy.
Link to the original article in Nature Communications:





