CMN Weekly (27 February 2026) - Your Weekly CRISPR...
CMN Weekly
Feb. 27, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...
On Friday 7th June, CRISPR Medicine News attended Retina International World Congress 2024. The meeting, hosted by Irish patient-led charity Fighting Blindness, united individuals affected by retinal disease, caregivers, clinicians, researchers, patient advocates and genetics counsellors from all over the world.
The theme ’Targeting the Cure’ highlighted the urgency in developing new treatments for the inherited retinal dystrophies (IRDs), a large group of genetic diseases that damage the retina, leading to progressive visual impairment and blindness.
To date, more than 250 IRD-causing genes have been identified, and while each of these might be rare on its own, IRDs as a group are actually among the most common genetic diseases and are estimated to affect one in 4,000 people globally.
The congress programme covered diverse topics from developing patient-care pathways to genotyping individuals living with an unsolved IRD, research advances in deciphering disease mechanisms and disease modelling, gene therapies, clinical trials, and diagnostics.
Of special interest to CRISPR Medicine News were oral presentations focused on gene editing by Mark Pennesi MD/PhD, a Professor of Ophthalmology from OHSU Casey Eye Institute and Director of Ophthalmic Genetics at the Retina Foundation of the Southwest in Dallas, TX, and Assistant Professor Leah Byrne PhD from University of Pittsburgh.

Besides following patients at Casey Eye Institute Ophthalmic Genetics Clinic, Mark Pennesi leads transitional research at OHSU and the Retina Foundation that he hopes will bring treatments for IRDs from the laboratory to the clinic.
At RIWC 2024, Mark presented a summary of the clinical data from Editas Medicine’s Phase 1/2 BRILLIANCE trial of EDIT-101 for the incurable disease Leber congenital amaurosis 10 (LCA10).
EDIT-101 is a CRISPR-Cas9-based therapeutic candidate designed to remove the intronic mutation IVS26 in the CEP290 gene, which accounts for almost one third of all LCA cases. This mutation leads to a premature stop codon in the CEP290 mRNA and a truncated, non-functional CEP290 protein. EDIT-101 comprises an adeno-associated virus serotype 5 (AAV5) vector containing a Cas9-encoding gene and two guide RNAs designed to simultaneously remove IVS26 and restore the CEP290 coding sequence.
LCA10 accounts for about 15-20 % of all Leber congenital amaurosis cases and arises through mutations in the CEP290 gene. The CEP290 protein plays a critical role in photoreceptor function, and loss-of-function mutations are associated with severe vision loss that is often present at birth. Remarkably, functional photoreceptors are present in some patients even when light perception is severely reduced, suggesting that it might be possible to restore some level of vision by correcting the CEP290 mutation(s) driving disease.
During the BRILLIANCE trial, EDIT-101 was administered to 4 cohorts comprising 12 adult and 2 paediatric participants, who either carry the IVS26 mutation on both CEP290 alleles (homozygous), or the IVS26 mutation on one allele and another deleterious mutation on the other CEP290 allele (compound heterozygote). In each patient, a single dose of EDIT-101 was delivered directly to the photoreceptor cells via sub-retinal injection.
Patients received a low, intermediate or high dose of EDIT-101 and were monitored for adverse events and toxicity as the key primary outcomes. The main secondary efficacy outcomes were measured using various tests for changes in baseline visual functions. The paediatric participants were followed for at least 6 months after dosing while some of the adult participants were followed for at least 2 years.
Six of the 14 participants exhibited a meaningful improvement from baseline in cone-mediated vision, which is the ability to see in bright light and to perceive colour.
When tested for visual acuity, which is the ability of the eye to distinguish shapes and details of objects at a distance, 4 of the 14 participants showed a significant improvement, with some patients able to see up to 10 lines on a letter chart.
A visual navigation challenge, which can be thought of as a simulated obstacle course, revealed improvements in navigation in 4 of the 14 participants.
When patient-reported outcomes were scored based on a questionnaire answered by the patients, 6 reported meaningful improvements in quality-of-life scores. In summary, 6 of the 14 participants experienced improvements in at least 2 of the parameters measured.
With respect to safety, adverse events, including inflammation and retinal deposits were reported in some patients, but according to the clinical data, these didn’t impact retinal function and resolved over time or with steroid treatment. In his closing remarks, Mark highlights that the functional improvements in this early trial involving severely impacted patients provide proof-of-concept that gene editing can work in vivo. Data from the BRILLIANCE trial was recently published in New England Journal of Medicine.
For reasons not related to the BRILLIANCE trial, Editas Medicine decided to suspend the trial after enrolment of the first 14 patients, and as of June 2024 it is unclear if EDIT-101 will undergo further clinical development.
Learn more about Mark Pennesi’s work at OHSU here.

As an Assistant Professor of Ophthalmology at University of Pittsburgh School of Medicine, Leah Byrne is leading efforts to characterise novel therapeutic targets for retinal diseases. At RIWC24, she presented some of her latest work in developing in vivo models of PRPF31 retinitis pigmentosa (RP), which is one of the more prevalent forms of dominant RP.
The PRPF31 gene encodes a splicing factor that is critical for mRNA processing. More than 100 deleterious mutations have been identified in the gene, and these lead to retinal degeneration through loss of photoreceptors.
The mutations behind PRPF31 RP, which is also known as RP11, cause disease through a phenomenon known as haploinsufficiency. This means that whether or not an individual heterozygous for mutated PRPF31 has the disease is determined by the expression levels of their healthy PRPF31 allele. Expression of the PRPF31 allele naturally varies by around 5-fold between individuals, which suggests that gene augmentation might a viable therapeutic strategy.
Retinitis pigmentosa (RP) refers to a group of rare, inherited, incurable retinal dystrophies that are associated with progressive vision loss due to degeneration of the light sensitive photoreceptor cells of the retina. Individuals are born with RP, with symptoms usually debuting in early childhood and worsening over time leading to blindness. RP is a complex set of diseases that can arise through mutations in more than 50 different genes, and disease severity varies greatly between affected individuals. Several inheritance patterns are known for RP, including autosomal dominant (30-40%), autosomal recessive (50-60%) and X-linked (5-15%).
To explore the possibility of treating RP11 by augmenting PRPF31 expression levels, Byrne and her team first developed a mouse model of the disease using an AAV vector expressing Cas9 and gRNAs to introduce various PRPF31 deletions. The genomic DNA of heterozygous mice was then characterised to identify the constructs that most efficiently knocked out PRPF31.
To target different cell types in the eye, three different delivery strategies were used, including sub-retinal injection, intravitreal injection (to target the centre of the eye), and systemic injection.
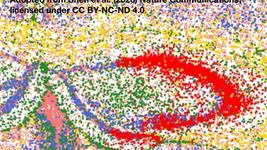
The team observed that sub-retinal injection of the gene-editing cargo led to efficient targeting of the photoreceptors at the back of the retina and better represented the human RP11 situation than intravitreal or systemic gene editing. They observed that 40-80 % of cells were edited for PRPF31 following sub-retinal injection, and this coincided with severe retinal degeneration seen by optical coherence tomography (a 3D scan of the eye). Further analysis using electroretinography to measure the electrical response of the photoreceptors indicated severely reduced retinal function in light and dark conditions, and immunostained cross sections of the impacted retinas revealed thinning and loss of photoreceptors, which is similar to the retinal morphology seen in RP patients.
Co-injection of the PRPF31 knockout construct and a Cas9-resistant construct containing a full-length wild-type PRPF31 sequence resulted in rescue of RP symptoms 5 weeks after treatment, and these improvements were even more pronounced 10 weeks after treatment. Here, eyes treated with both constructs were protected from degeneration as seen via 3D scanning, retinal thickness measurements and heat maps, which showed that retinal protection was most pronounced around the injection site. These data support PRPF31 gene augmentation as a therapeutic approach for RP11 and have been published in Nature Communications.
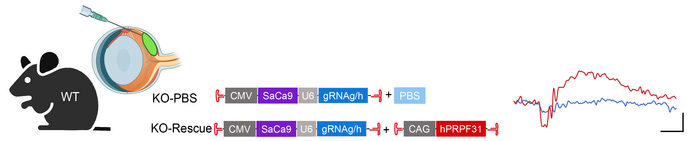
The team are now focused on translating their findings in proof-of-concept studies in a primate model of RP11. During Leah’s conclusion, she highlighted that the CRISPR models used so far involve editing of both PRPF31 genes which doesn’t mimic the haploinsufficiency seen in humans with RP11. To address this limitation, her team is also working to develop animal models that can recapitulate haploinsufficiency.
Read more about Leah Byrne’s work at University of Pittsburgh here.
Fighting Blindness is an Irish patient-led charity with a mission to cure blindness, through funding research into new treatments and raising awareness about degenerative retinal diseases. To date, the charity has invested over €20 million in more than 115 research and clinical projects. Read more about Fighting Blindness here.
Read more about RIWC24 here.
To get more CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.
ArticleNewsLeber Congenital AmaurosisRetinitis PigmentosaEditas Medicine, Inc.