FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

The enormous clinical potential of CRISPR comes with a price tag: most gene-editing therapies require a highly personalised and, therefore, costly approach. Moreover, even CRISPR medicines applicable to larger groups of patients are likely to be very expensive. So how can society regulate the clinical practice of gene editing to ensure safety while also keeping astronomical costs at bay?
In search of answers, CRISPR Medicine News talks with Dr Tessel Rigter and Professor Martina Cornel from Amsterdam UMC. Both scientists work at the interface of clinical genetics research and implementation of genomic and societal insights into standard healthcare.
»Each year, approximately 250,000 babies are born with sickle cell anaemia globally,« says Martina Cornel.
»Gene editing using CRISPR offers a potential cure for these affected individuals and holds promise as an effective treatment for many more rare genetic diseases. As such, we are anticipating a ‘tsunami’ of prospective gene-editing therapies.«
Around the world, various regulatory agencies like the European Medicines Agency (EMA) in the European Union, and the Food and Drug Administration (FDA) in the United States, regulate medicines as finalised products. In contrast, gene-editing therapies need to be optimised for individual patients to target unique DNA changes. Unfortunately, the current systems regulating drugs are not suited to regulate the safety and effectiveness of these highly personalised clinical gene-editing applications, especially for smaller groups of patients, argue Cornel and Rigter.
»Besides the lack of clear regulatory paths to market for clinical gene editing, there are also questions concerning treatment availability, costs, and reimbursement,« Rigter adds.
Healthcare strategists recently estimated that the research and development costs of a new molecular-based treatment for an orphan disease might amount to half a billion US dollars. Traditionally, pharmaceutical companies organise costly randomised clinical trials to obtain regulatory approval for medication in development. This road to market may be feasible for genetic conditions involving larger groups of patients, like some of the current CRISPR approaches to treat sickle cell anaemia. However, CRISPR medicines for rare genetic diseases that may only apply to one specific patient make randomised clinical trials impossible and present a lack of financial incentives for the pharmaceutical industry to develop these highly personalised therapies.
“It is not feasible, nor cost effective, to perform trials and scrutinise safety for every unique gene deficiency that you want to correct, which basically is the current regulatory practice”Tessel Rigter
»It is not feasible, nor cost effective, to perform trials and scrutinise safety for every unique gene deficiency that you want to correct, which basically is the current regulatory practice,« Tessel Rigter says.
Professor Cornel adds: »We are heading into unknown territory regarding the implementation of gene-editing medicines. Current licensing and reimbursement systems are inadequate. We need to implement new regulatory measures to ensure safe and affordable gene-editing treatments. Worldwide, professionals are grappling with this disparity, pitching ideas and searching for solutions to bridge this gap. And don’t forget that, despite the enormous potential of gene editing medicines, we still need actual proof of safety, effectiveness and clear clinical benefits. It is still very early days regarding the validation of clinical CRISPR efficacy.«
Current fast-paced clinical trials present challenges in evaluating long-term effects, and there is the potential that severe long-term complications might offset benefits. Despite these unknowns, laying the groundwork for implementing CRISPR human gene-editing in the clinic is needed now, as developing regulation is a slow process and subject to wider public debate.
Martina Cornel and Tessel Rigter recently published an overview of how CRISPR treatments might navigate the current regulatory practices in the EU. Unfortunately, despite some flexibility, these routes do not suffice when personalised CRISPR medicines become available for more patients and many monogenetic diseases.
Cornel and Rigter point to the rollout of another new and costly personalised treatment to shape the outlines of CRISPR medicine's road to market. CAR T-cell therapy involves programming a patient's own immune cells to recognise and destroy blood and bone marrow-derived cancer.
In the United States, the FDA created new programmes to streamline the regulatory processes for developing drugs like CAR T. In the EU, the pharmaceutical companies involved in CAR T-cell production are required to provide educational programs for healthcare professionals and patients in compliance with national regulatory authorities. Furthermore, the number of national treatment centres are currently limited to foster a rapid gain in expertise. The high costs of CAR T-cell treatments are presently restricting availability for patients. Still, ongoing technological innovations like 'off-the-shelf' CAR T-cell therapies are expected to reduce expenses drastically in the future. A similar trajectory is likely with gene-editing therapies.
Future regulations need to balance scientific data that support clinical safety and efficacy, costs, and treatment availability.
»We are envisioning a new way to authorise and make CRISPR-based medicines available to patients. We call it the 'Bio-Nespresso model',« says Martina Cornel.
If you are familiar with a Nespresso coffee maker, you can appreciate that the hardware - the coffeemaker - is the same for each cup of coffee that is made. Nevertheless, the standard equipment brews coffee in distinct flavours, tailored to individual taste, simply by providing the machine with the suitable capsule holding the desired coffee.
»In CRISPR terms the authorities can carefully regulate oversight and safety at the level of the 'coffeemaker', that is the clinic and training of medical staff plus production standards for shared features like delivery systems, the CRISPR enzyme, and for example, cell culture conditions for ex vivo treatments,« says Cornel.
»However, when it comes to the 'content of the capsule', regulations have to provide freedom to use customised guide-RNA sequences or potential DNA templates to address the needs of all the different patients with monogenetic diseases.«
Dr Kirmo Wartiovaara, a specialist in clinical genetics at the University of Helsinki in Finland, offers surgery as another analogy to implement somatic gene editing, ESPECIALLY for rare diseases.
»In surgery, machines and instruments are regulated, but then it’s up to the qualified staff how to use them. Each operation is somewhat different, best approaches are published and patients sign hospital consent forms.«
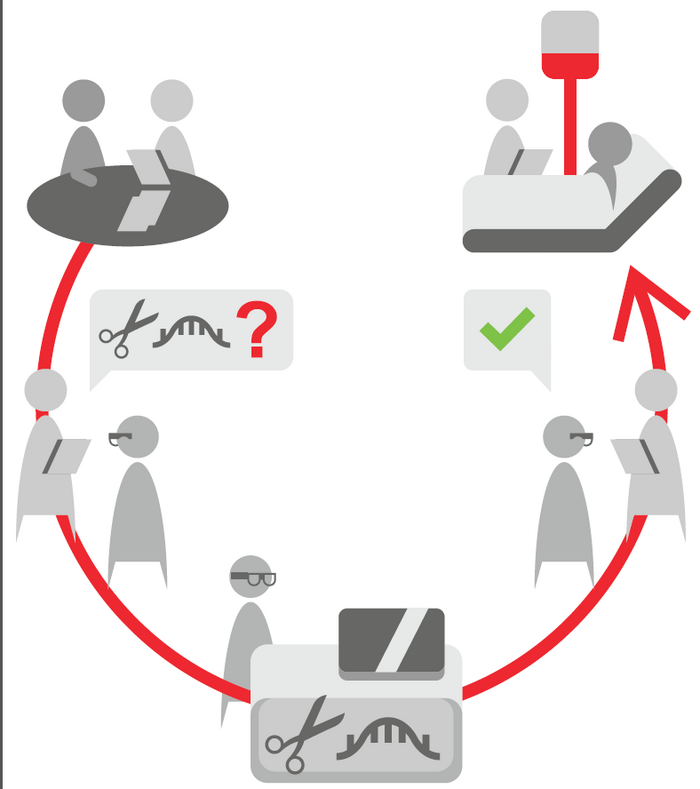
To implement a system such as the 'Bio-Nespresso model' will likely involve a close collaborative effort between regulatory agencies, clinical expert centres at public research institutes, patient groups, and private pharmaceutical companies, according to Martina Cornel.
In the US, a recent collaborative initiative sponsored by the National Institutes of Health (NIH), the Somatic Cell Genome Editing (SCGE) Consortium, was established to accelerate the translation of safe and effective genome-editing therapeutics into the clinic.
Public research institutes and pharmaceutical companies are working together to collect data and build an SCGE toolkit composed of new genome editors, delivery technologies, and biological systems, including animal models and primary human cell lines and organoid platforms. Importantly, these tools will be available for distribution to the wider biomedical community.
The safety and efficacy data from early clinical trials administering CRISPR medicine will be critical for further expanding gene-editing therapies. The balance of costs and patient benefits will be essential, as well.
»It is neither feasible nor cost-effective to perform trials and scrutinise safety for every unique gene deficiency that you want to correct, which basically is the current regulatory practice. Also, the more safety measures you implement, the more data is required from clinical trials, the higher the final product cost will be, the less likely industry will invest in the development of these products”« says Tessel Rigter.
Fortunately, there is no apparent need to develop the entire CRISPR therapy for each unique patient from scratch.
The specific mutation in the affected gene will have to be addressed by designing unique guide RNAs and correction strategies for many monogenetic diseases.
However, in line with the Bio-Nespresso model, the delivery and enzyme technology will apply to groups of patients with shared disease features despite distinct genetic causes. For example, the CRISPR technology developed to treat sickle cell anaemia targets haematological stem cells. Therefore, the same clinical ex vivo strategy for CRISPR delivery and function can apply to other monogenetic blood diseases like beta-thalassemia, severe combined immunodeficiency, or congenital anaemias.
Similarly, it seems feasible to group patients with hereditary metabolic diseases under one CRISPR strategy targeting the liver or implementing a standard CRISPR protocol to treat patients with distinct disorders affecting the central nervous system. Given the rarity of many monogenetic diseases, such an approach will increase the patient numbers treated under a common protocol and boost therapy availability due to a lower development cost per unique patient.
CRISPR medicines will likely be administered to patients in a limited number of qualified clinical centres with specially trained medical professionals, similar to the implementation of CAR T-cell therapies.
»We foresee that oversight on safety and efficacy with CRISPR therapies involving unique patients will be organised via clinical genetics departments affiliated with the designated clinical CRISPR expertise centres,« says Martina Cornel.
»As part of care provisions, clinical geneticists may follow treated patients over time and inform regulating authorities, peers, and patient organisations all connected in international networks about therapy efficacy, observed complications, or side effects.«
She realises it will take an extra effort but at the same time points to the COVID-19 pandemic for inspiration.
“When you want to innovate, you have to make investments to facilitate these innovations”Martina Cornel
»When there is a sense of urgency, the development of new therapies can happen quickly. That's what we have seen in the SARS-CoV-2 pandemic. A concerted effort by governments, public institutions, and private companies pushed novel technology forward and produced effective COVID-19 vaccines in record time,« Cornel says.
»Why don't we organise a concerted effort to rapidly develop gene-editing medicines for many orphan diseases with an unmet clinical need? There is another type of urgency here, and when you want to innovate, you have to make investments to facilitate these innovations.«
Henri van de Vrugt is a molecular biologist and Chief Scientific Officer at New Haven Biosciences Consulting, US.





