
Intellia Files BLA After Lonvo-z Meets Phase 3...
CMN Clinical
Apr. 29, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Ever since researchers first started to use Cas9 and other programmable nucleases to perform precisely targeted gene editing, they found themselves in need of tools to predict and identify the outcome of their experiments. One of the pioneers who have tried to fill in that knowledge gap is Associate Professor Eric Paul Bennett at the University of Copenhagen.
Bennett invented one such tool - Indel Detection by Amplicon Analysis (IDAA) - in 2015. Recently, he took it one step further with a comprehensive review of what he calls the ‘Achilles heel’ of precise genome editing. The central problem is that even though we now have a plethora of methods to detect and predict the outcome of gene editing experiments, we need standardisation in the approach, so that researchers can learn from as well as compare their findings to the work of others.
»The current status is that anybody - all labs or pharma companies - has their own way to do both the genome editing and identify the outcomes. So this makes it very difficult to compare results between labs and between companies because the workflows and the methods are completely different,« Bennett says.
The cornerstone of Cas9 and other programmable nucleases (PNs) is their ability to cause DNA double-strand breaks (DSB) at target sequences determined by either a guide RNA (gRNA) or direct DNA binding activity of the nuclease. DSBs may also occur naturally by, e.g. UV light, ionising irradiation or metabolic byproducts, and in any case, the cell will try to repair the break by joining the cut ends.
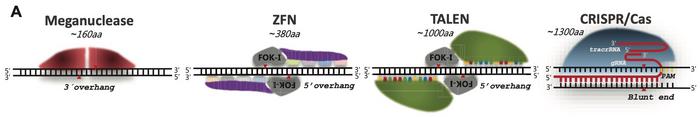
The review starts by outlining how this can occur by four main pathways that result in either perfect repair or formation of so-called indels, i.e. small insertions or deletions. An essential consideration when it comes to indels is how many nucleotides have been deleted or inserted during the DNA repair process. If this number is divisible by 3, an in-frame mutation will occur, which will result in a protein with altered structure and potentially also altered function. If, on the other hand, this number is not divisible by 3, an out of frame mutation will occur, resulting in a truncated and most often non-functional protein, i.e., a so-called knock-out mutation.
The most common repair mechanisms are non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ). NHEJ is unique in being the only repair mechanism that can lead to insertions, and the most common indels induced by Cas9 are indeed 1-bp insertions, accounting for 10–25% of all events. The 1-2 bp deletions and >2-bp insertions collectively account for 20-25% and 30-40%, respectively, of all indels. While NHEJ only performs small insertions of up to 4 bp, MMEJ is responsible for most 2-30 bp insertions.
The kind of repair mechanism and the specific indels introduced after Cas9 or another PN have made a DSB depend on many factors. Cellular proteins required for each repair mechanism might not be available in certain cell cycle phases; NHEJ is active in all phases except for mitosis, while MMEJ is restricted to S and G2 phases. Moreover, NHEJ tends to occur within a few hours after DSB induction, whereas MMEJ takes place only after 1-3 days.
Availability of necessary cellular proteins might also depend on cell type and whether the cell is cancerous. This will influence the repair mechanism being used and thus the nature of the indels introduced. Additional factors such as characteristics of the gRNAs, chromatin structure at the target site, and the delivery method used can also affect the outcome of gene-editing experiments. To make things even more complicated, the overall outcome will also be quite different in individual cells of a sample.
“We need experiments to verify that a gRNA also behaves as predicted, but a good algorithm will give a first-hand selection criteria for avoiding ones that will not give the desired outcome”Eric Paul Bennett
Research has shown that the relative frequencies of predominant indels for a given Cas9:gRNA pair are quite similar between replicate experiments performed in the same cell type and under the same conditions. However, it is still difficult to predict outcomes when experiments are performed under slightly different conditions. Attempts to build algorithms that predict indel formation from various gRNA designs are thus limited by the model experiment’s premise, e.g., delivery method, cell type, and target sites for the experimental gRNAs.
Accordingly, different prediction models have shown significant differences when compared side-by-side, and many gRNAs with a high predicted activity have been found in actual experiments to be inactive. Bennett says:
»We need experiments to verify that a gRNA also behaves as predicted, but a good algorithm will give a first-hand selection criterion for avoiding ones that will not give the desired outcome.«
Fortunately, researchers can choose from several methods to analyse the nature of the indels they have achieved. All methods use PCR to initially amplify the target site where indel formation has occurred, and subsequently, several methods can be employed to analyse the amplicon. The methods differ considerably in terms of effort required, time and cost effectiveness, and their resolution and sensitivity of indel detection.
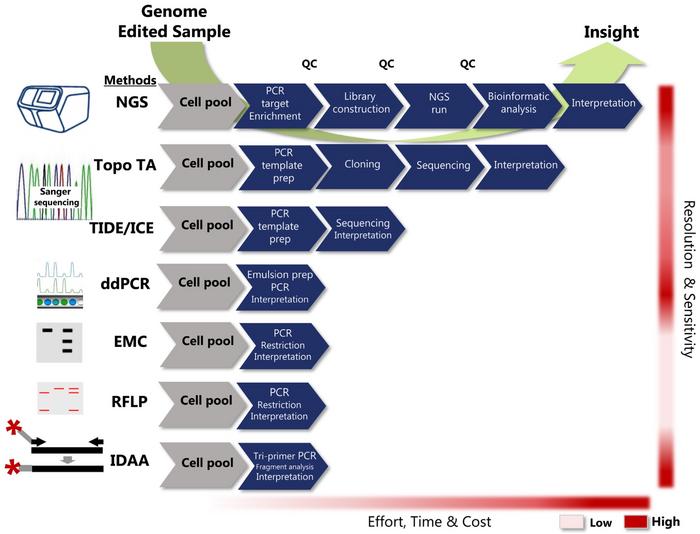
Some methods will give you the sequences of the edited target sites and reveal the indels’ precise nature, while other techniques will only tell you if there has been some edit or not. Exactly what information is needed from an indel detection strategy depends on the purpose of the given experiment.
»About 80% of researchers using CRISPR are making knockouts by out of frame deletions. In that case, you just want to determine if you have an out of frame-causing indel, and excessive analysis to detect the exact nature of the indel by next-generation sequencing might be overkill,« explains Bennett.
Bennett started to utilise CRISPR in its very early days within the field of glycobiology. He found himself missing a high throughput, cost-efficient and high-resolution assay that could detect indels with the same or similar sensitivity as next-generation sequencing (NGS). He explains:
»To suit that need, we developed the Indel Detection by Amplicon Analysis (IDAA) assay, where you actually utilise the power of the DNA sequenator instrument to get single base resolution of PCR products. The trick is to use a fluorophore-labelled universal primer and a tri-primer setup to label PCR products homogeneously. The amplicons are then separated by capillary electrophoresis and detected and quantified as peaks based on size and fluorescence intensity.«
IDAA can provide a quantitative determination of indels in complex editing profiles with single-base discrimination. Moreover, it has an indel detection sensitivity down to 0.1%, i.e. similar to NGS, and a turn-around time from sample to a full insight in less than 6 hours.
For clinical use of CRISPR and other gene-editing technologies, the lack of robust algorithms for indel prediction and indel detection methods is critical. This applies especially to in vivo gene-editing therapy development where the edit cannot be undone if it turns out not to be as expected. It is challenging to compare results between labs and companies because they might use entirely different workflows and methods that affect indel formation outcomes in distinct ways. This will ultimately challenge the FDA, EMA, and other regulatory bodies when they will have to evaluate data from clinical trials of gene-editing therapies in the coming years.
“I think this is a very important step to harmonizing the way we detect the outcome of genome editing”Eric Paul Bennett
»One consortium that is addressing this issue is the National Institute of Standards and Technology (NIST) Genome Editing Consortium,« says Bennett, who explains that NIST will make sets of standardised DNA samples containing known indels, and provide these to scientists so they can adjust their own methods to arrive at the same result.
»The results of these efforts will come out in the next few years, and we will see how comparable the different methods are. So NGS, bioinformatics, deconvolution and all that has to come together and hopefully show similar results on those particular standards. I think this is a very important step to harmonising the way we detect the outcome of genome editing,« Bennett says hopefully.
Link to the original article in Nucleic Acids Research:
https://pubmed.ncbi.nlm.nih.gov/33170255/
ArticleNewsOff-targetCRISPR-CasMeganucleasesPrime editorsTALENsZFN - Zinc finger nucleases





