FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...
Exciting new clinical data lend further support to CRISPR-Cas9 gene editing as a potential cure for two devastating, incurable disorder that afflicts millions of people around the world - transfusion-dependent β-thalassemia (TDT) and sickle cell disease (SCD), respectively.
The data presented at the 25th Annual European Hematology Association (EHA) Congress demonstrate how three patients are symptom-free. The two TDT patients no longer need regular blood transfusions up to 5 and 15 months respectively after the treatment began, and the SCD patient is free of vaso-occlusive crises at 9 months.
»The data announced today are remarkable, including the demonstration of clinical proof-of-concept in TDT,« said Reshma Kewalramani, CEO and President of Vertex Pharmaceuticals in a statement.
They are developing the treatment called CTX001 jointly with CRISPR Therapeutics.
Samarth Kulkarni, CEO of CRISPR Therapeutics stated: »These highly encouraging early data represent one more step toward delivering on the promise and potential of CRISPR-Cas9 therapies as a new class of potentially transformative medicines to treat serious diseases.«
The data has been eagerly anticipated since November 2019 when the first preliminary data was announced on a US patient treated for SCD and a TDT patient treated in Germany.
Now there is longer-duration follow-up data on these first patients as well as a second TDT patient, who is transfusion independent at 5 months after beginning the treatment. Also three more TDT patients and one SCD patient have started therapy for a total of seven patients.
The trials are called CLIMB-111, and CLIMB-121 for TDT and SCD, respectively, and both are phase 1/2 safety and efficacy trials, that each aim to enrol up to 45 patients.
The cause: A mutation in a gene that codes for the β-subunit of haemoglobin - the iron-containing protein in red blood cells that carries oxygen to cells throughout the body.
β-thalassemia patients don't make enough haemoglobin, which leads to anaemia and fatigue, and patients with the most severe form, β-thalassemia major, rely on monthly red blood cell transfusions to survive.
In sickle cell disease patients, the abnormal form of haemoglobin distorts the usually flexible and doughnut-shaped cells to resemble a sickle shape. The sickled cells can clump together and obstruct small vessels, cause organ injury, and pain crises.
Both diseases can lead to early death.
Both blood disorders are caused by mutation of a gene that codes for the β-subunit of haemoglobin - the iron-containing protein in red blood cells that carries oxygen to cells throughout the body.
The CTX001 investigational therapy uses CRISPR-Cas9 genome editing in a clever workaround that works for both diseases.
Instead of using CRISPR to edit the actual mutations (of which more than 200 are known for TDT), the therapy edits a patient’s haematopoietic stem cells with the aim of reactivating the production of fetal haemoglobin (HbF) which is thought to substitute for diseased haemoglobin in TDT and SCD patients, potentially reducing or eliminating symptoms..
This entails harvesting blood stem cells from the patients, editing them in the lab, and reinfusing billions of them back into the patient. The patients have to undergo chemotherapy to remove their remaining defective stem cells and make room for the edited cells to establish a permanent supply of healthy blood cells.
Now the data for the first patients looks very encouraging.
The first step to populate the bone marrow and begin to produce the mature blood cells, a process that is known as engraftment, has been successful for all seven patients, and take about 30 days.
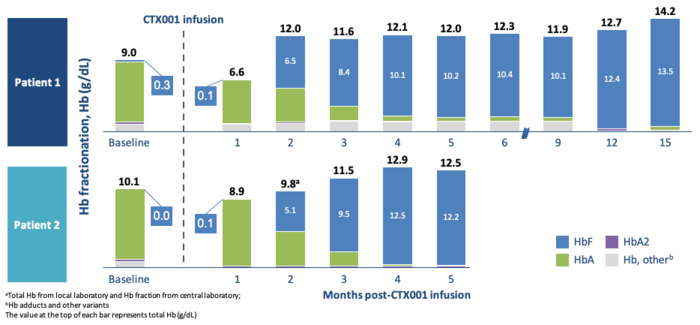
For the two β-thalassemia patients, the level of HbF rose quickly after engraftment to reach healthy levels of haemoglobin in just three months. For both patients, almost all haemoglobin was HgF and none HgA. (See figure above).
At the final time point (15 and 5 months), the patients had haemoglobin levels of 14.2 and 12.5 grams per deciliter, respectively, compared to a range of 12 to 15 normally seen in healthy adults.
The treatment has had a substantial impact on the lives of the patients. Both have become transfusion independent - before the treatment, they had transfusions of 34 and 61 units of packed red blood cells per year, respectively.
The CTX001 results demonstrate proof of concept in patients with TDT, according to Vertex. Though it is still early, the extended 15-month follow-up data support the durability of the treatment effect.
“These are remarkable and unprecedented results”
»These are remarkable and unprecedented results,« a Vertex spokesperson said in an email.
»We demonstrated that we could take CD34 cells from patients, edit them ex vivo with CRISPR-Cas9, re-infuse the edited cells into patients, achieve engraftment, and then show a clinically meaningful increase in HbF and total Hb, that has been maintained for up to 15 months. Also, we demonstrated clinical benefit with transfusion independence in two TDT patients and no vaso-occlusive crises in one SCD patient. And we did this safely.«
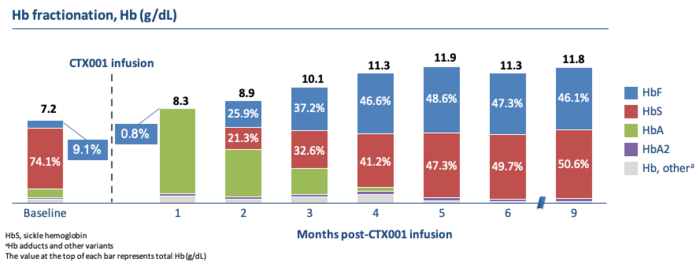
The sickle cell patient, Victoria Gray, who has given a 1st anniversary interview, also reached and maintained healthy haemoglobin levels of almost 12 grams per deciliter. (See figure above).
Unlike the TDT patients, her haemoglobin composition was about half of HbF and HbS (the sickling haemoglobin) each.
Importantly after nine months, Victoria Gray is transfusion independent and has remained free of the painful complications known as vaso-occlusive crises (VOC), which require hospitalization. She previously suffered VOC seven times per year.
It is also reported that all three patients have experienced severe adverse events during the treatment, such as pneumonia. However, none of the events was attributed to CTX001 therapy.
Asked about off-target concerns Vertex replied via email that they have sequenced thousands of potential sites across the genome for off-target cutting and seen no off-target effects.
The success rate of editing was also monitored. The researchers measured the allelic editing before infusing the edited blood stem cells back into the patient, and continue to monitor this in the peripheral blood, and the bone marrow periodically during the study.
The important measure for patients is the allelic editing in the bone marrow after the procedure, which were high - above 75% - in both the first TDT and SCD patient at 6 months following infusion of CTX001. And for the first TDT patient the data show the allelic editing is still above 75% past 12 months.
»We believe this metric demonstrates the potential long-term efficacy of CTX001,« the Vertex spokesperson wrote in the email.
Even though the trials plan to enrol up to 90 patients, and thus it is still early days, the results so far support CRISPR-Cas9 gene editing as a functional cure for these devastating diseases.
»We are still early in the development of CTX001, but we believe that having a potential one-time curative therapy for this serious disease would be a very compelling treatment option for many TDT and SCD patients, especially for those with severe disease who have multiple severe pain crises and hospitalizations each year,« the Vertex spokesperson wrote.
CTX001 is far from alone, and multiple other trials and strategies are underway.
For example, Sangamo Therapeutics, partnered with Sanofi, is also testing the strategy of unlocking fetal haemoglobin in blood stem cells of TDT and SCD patients. Sangamo uses zinc fingers nucleases (ZFN) for the genome editing instead of CRISPR and announced positive early data in 2019, but unlike CTX001, these patients were not transfusion independent.
Another example is Editas Medicine, which just announced pre-clinical data for their EDIT-301 to treat SCD. The treatment also entails harvesting and CRISPR-editing the blood stem cells, but the strategy is a little different. They focus on the HBG1/2 promoter in the β-globin locus to induce HbF and use CRISPR-Cas12a to copy naturally occurring HbF-inducing mutations. EDIT-301 hasn't advanced to a clinical study in humans yet but plans to begin clinical testing later in 2020. Editas believes that its approach is superior to the ones being taken by Vertex/CRISPR Therapeutics and Sangamo and aim for 'best-in-class'.
The main competition is likely to be a gene therapy by bluebird bio. On the same day as the Vertex/CRISPR Therapeutics announcement bluebird bio announced findings from its phase III β-thalassemia gene therapy clinical trial. They show that a majority of patients across a range of genotypes achieve transfusion independence with haemoglobin levels that are near-normal (≥10.5 g/dL). The bluebird gene therapy is a one-time treatment adding functional copies of a modified form of the β-globin gene into a patient’s blood stem cells. After successful treatment, the patients produce the gene therapy-derived haemoglobin at levels that eliminate or significantly reduce the need for transfusions.
For SCD and TDT patients these are good news raising hope of not just one but several potential one-time cures.
It is also exciting for the broader gene editing field as these treatments may very well pave the way to treat other hard to treat heritable diseases.
With the CRISPR-Cas9 gene-editing tool, one obvious fix would be to edit the disease mutation. However, the new CTX001 therapy uses a clever workaround, and instead aim to increase the level of a haemoglobin form called fetal haemoglobin (HbF). Why we have two forms of haemoglobin is not known, but shortly after birth, babies switch from the fetal to the adult form (HbA).
The strategy is based on observations of rare individuals that continue to make HbF throughout their lives and stay healthy. An even more rare subgroup of these individuals also has SCD or β-thalassemia. Still, these individuals only show extremely mild symptoms because the high level of HbF protects against the disease. The idea is to mimic these variants.
More than a decade ago, it was discovered that a transcription factor called BCL11A is needed to turn off the production of fetal haemoglobin in the red and mediate the switch to adult haemoglobin. And later it was found that without a functioning BCL11A, the red blood cells will produce the HbF.
The CTX001 therapy uses CRISPR-Cas9 to make a small deletion in the BCL11A gene and knock out the protein in blood stem cells harvested from the patients. The edited stem cells are then infused back into the patient to establish a permanent supply of healthy red blood cells producing fetal haemoglobin.
ArticleNewsSickle Cell Disease, SCDTransfusion-Dependent Beta Thalassemia, TDTGene therapyCas9Clinical





