FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Spearheaded by collaborators Dr. Wenzhen Duan, Professor of Psychiatry and Behavioural Science at Johns Hopkins University School of Medicine, and Dr. Gene Yeo, Professor of Cellular and Molecular Medicine at the University of California San Diego, a new proof-of-concept study has demonstrated that Huntington’s disease (HD) phenotypes can be rescued in patient cells and an in vivo mouse model of HD.
Their recent Nature Neuroscience publication describes the use of Cas13d to deplete the mutant mRNA transcripts that contribute to the pathogenesis of HD. Experiments in mice showed substantial benefit of the therapeutic approach, including significantly improved motor function.
While therapies targeting mRNA transcripts are typically expected to have only short-term effects, Duan, an expert in HD and other neurodegenerative disorders, explains that their results appear to have real staying power:
»A single injection of the therapy in mice shows rescue of the disease phenotype for at least eight months.«
HD is a late-onset, fatal neurodegenerative disorder with no cure; currently available treatments merely provide modest relief of symptoms. HD is caused by a gain-of-function mutation: a trinucleotide repeat hyperexpansion in the first intron of the HTT gene, which codes for the Huntingtin protein.
Similar to Friedreich’s ataxia, another neurodegenerative disease caused by a trinucleotide repeat hyperexpansion, the number of repeats in the expansion is inversely correlated with the age of onset of disease symptoms. Inherited in an autosomal dominant manner, the mutation causes toxic Huntingtin protein to be produced. Brain shrinkage results in cognitive decline and movement disorders, and death within 20 years of clinical onset.
»For diseases like this, it’s fatal and there is no other approach, and the patients really welcome any possible option. So it’s actually relatively easy to recruit Huntington’s patients for clinical trials – they want preventative treatment, because they know they will definitely develop severe disease,« Duan says.
Duan and Yeo’s approach was to deplete mutant HTT (mHTT) transcripts using CRISPR-Cas13d, which targets and cuts RNA. While it’s not a permanent cure, there are significant reasons for their choice of an RNA-targeting approach for a disease like HD. Yeo, a computational biologist and expert in RNA-targeting, RNA biology and neuroscience, explains the rationale for not using a CRISPR-Cas9 approach for genome editing:
»I’m very supportive of a DNA-targeting approach, but we’re not there yet, at least for central nervous system disorders. DNA repair does not happen efficiently in neurons.«
Other studies have attempted to use RNA interference (RNAi) and antisense oligonucleotides (ASO) to the same end with some pre-clinical success. However, these methods have so far been unable to specifically target the mutant allele, preventing their clinical translation. Since HD patients need the wild-type HTT allele to be intact for key CNS functions, the Cas13d method developed by Duan and Yeo was designed to selectively target the CAG hyperexpansion (CAGEX) and eliminate only the mutant transcripts.
Yeo’s team designed and tested the Cas13d-CAGEX system in induced pluripotent stem cells (iPSCs) derived from HD patient cells, with each cell line featuring a variable number of CAG repeats. The results showed significant depletion of mHTT transcripts, between 56.2 and 84.1%. HD-associated molecular phenotypes – differentially expressed genes involved in HD-mediated pathology – were also reversed in the treated cells, which Duan and Yeo suggest is a useful measure of the therapy’s efficacy.
Professor Robert Lahue of the University of Galway’s Centre for Chromosome Biology, an expert in trinucleotide repeat instability who did not contribute to the study, commented on the significance of the team’s approach.
»One of the broader implications is the possibility of applying this technology to mRNA in other diseases to eliminate a toxic protein or RNA molecule. In principle, the approach has applications to a number of genetic disorders,« Lahue remarks.
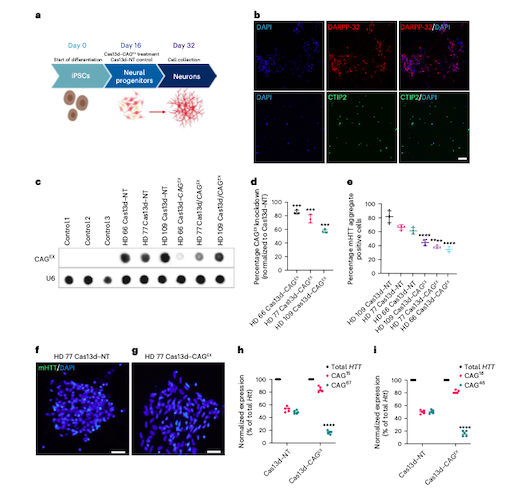
Figure 1: Cas13d–CAGEX reduces mHTT mRNA and protein in cells derived from patients with HD. (A) Schematic of the differentiation protocol used for iPSC-derived neurons treated with Cas13d–CAGEX. (B) Representative immunofluorescence images of cells at day 32 in control neuronal cells, showing that the cells are positive for striatal markers, DARPP-32 (top) and CTIP2 (bottom). (C, D) RNA dot blot analysis and quantification of CAG-expanded RNA within MSN cells transduced with Cas13d–NT- or Cas13d–CAGEX-expressing lentiviral vectors (E-G) Quantification of mHTT aggregates within MSN cells transduced with Cas13d–NT- or Cas13d–CAGEX-expressing lentiviral vectors (H, I) Quantification of mutant and WT HTT RNA via allele-specific RT-qPCR in GM04723 and GM02151 fibroblasts. Morelli et al. Nature Neuroscience 2022 https://doi.org/10.1038/s41593-022-01207-1
Cas13 systems gained some early notoriety for their collateral activity, where once it has recognised and cut the target transcript, Cas13 can also cut all other transcripts within the cell, often causing cell death. While the collateral activity of Cas13 systems is still poorly understood and requires more study, Cas13d is a variant that appears to have less cause for concern, and the team’s experiments using Cas13d in human cells showed that collateral activity was minimal.
»We didn’t see a large collateral effect, and we think it’s an expression-dependent feature of the Cas13d protein. It may be that the expanded repeat traps the protein in a certain way that prevents it from destroying other RNAs. What we’re trying to do now is leverage variation in the Cas13 protein that decouple the initial target recognition and cleavage from the collateral conformation - the ternary complex. We’re also exploring other proteins and systems for targeting RNA with less collateral activity,« Yeo says.
Following on from positive results in cells, Duan’s team then tested the therapy in vivo in a knock-in mouse model of HD, packaging Cas13d and the sgRNA into an adeno-associated virus (AAV) and delivering via intra-striatal injection. Treated HD mice showed significantly improved motor function which lasted up to eight months.
The current approach requires neurosurgery to get the therapy to the affected neuronal cells and uses an AAV vector. While this is feasible in human patients, it’s not ideal, and the team would prefer to move away from an AAV-based approach if possible.
»Our biggest challenge is delivery. My lab is collaborating with biomedical engineers to develop different methods for delivery, such as lipid nanoparticles and exosomes,« Duan elaborates.
In terms of next steps, Duan and Yeo are committed to safety, and they continue to work together in the hope they can eventually push the therapy towards the clinic.
»We still need to do longer-term mouse studies, and check for immunogenicity and other side effects. Then we will move on to a larger animal model. HD therapeutics are often tested in pig models, because the brain is the same size, and knock-in pig models for HD are available,« Duan notes.
While using CRISPR-Cas9 to excise the CAG repeat in the HTT gene would be preferable because it offers a permanent cure, the success of the study still offers considerable hope to the HD patient community, as Lahue concludes:
»One of the surprises in the study is how long the test therapy lasted in mice. Eight months is a significant portion of the mouse lifespan. Targeting the transcripts using CRISPR-Cas13d could help buy precious time for patients, their families, medical professionals, and researchers until it becomes clear if gene editing will be feasible, safe and efficacious in HD.«
To get more of the CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.
Rebecca Roberts is a molecular biologist and science writer/communicator based in Queensland, Australia.
ArticleInterviewNewsHuntington's diseaseOther Genetic ConditionsCas13





