CMN Weekly (20 February 2026) - Your Weekly CRISPR...
CMN Weekly
Feb. 20, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

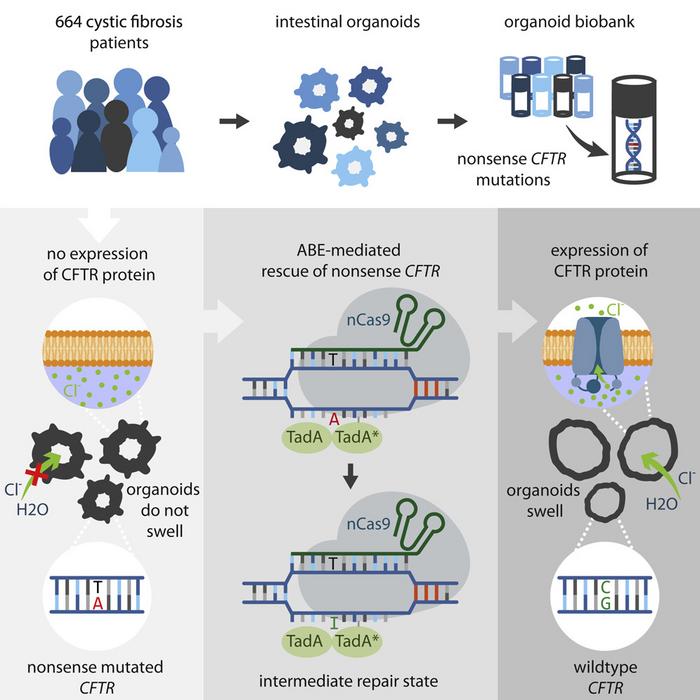
Dutch scientist Hans Clevers and colleagues in Utrecht have cured cystic fibrosis in mini-organs grown from patient stem cells. Using the newly developed CRISPR-tool called base editors, they remove stop-codon mutations in the cystic fibrosis gene and turn it back into the original amino acid.
»I have never seen anything like CRISPR. Everything we try works. It's amazing,« says Hans Clevers, at the Hubrecht Institute, Utrecht University in the Netherlands.
On top of the editing success, the researchers found no evidence of off-target effects, which is one of the biggest safety concerns surrounding the use of CRISPR for gene therapy.
The research could pave the way for gene therapy based on two of the most promising new technologies CRISPR and mini-organs.
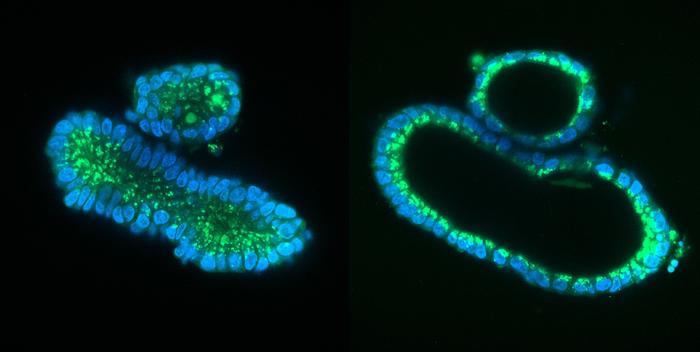
Mini-organs have attracted a lot of attention in the last decade, but what are they? Briefly, mini-organs or organoids are small pencil tip-sized lab-grown versions of organs, that are functionally similar and replete with the major cell types. The organoids can be grown from stem cells found in small tissue samples with the right nutrition and growth factors - 'the secret sauce' as Clevers calls it. Hans Clevers identified a cell in the gut that can give birth to all other intestinal cells and grew the first 'mini-guts' over a decade ago.
Collaborator Jeffrey Beekman at Wilhelmina Children's Hospital in Utrecht had the idea to develop the mini-guts into a cystic fibrosis functional assay.
The assay is currently used in the dutch health care system to predict if a costly drug called Orkambi by Vertex Pharmaceuticals will have an effect in individual patients.
»If you have a positive organoid test, you get the drug and so far it has always worked,« says Hans Clevers.
The problem is that it is challenging to predict if the drug will or will not work since many patients have a different version - cystic fibrosis can arise from over 2.000 mutations in just one gene, and many patients have rare or even unique mutations.
Over the years, the researchers have built a biobank that now holds mini-guts from 664 dutch cystic fibrosis patients - almost half of the country's cystic fibrosis population.
Having this unique resource Clevers and colleagues thought it would be ideal for studying if the latest CRISPR technology could be used to correct the mutations safely in human cells.
The so-called base editors were developed in 2018 and are Cas9 fusion proteins that can be guided to a specific sequence in the DNA and change one base-pair to a different base-pair. There are not base editors for all combinations but A-T to a G-C and vice versa exists and going through the biobank it turns out that about 20% of the patients could, in theory, be cured using the base editors. Importantly this includes patients with stop codon mutations for whom there currently is no medication.
It is a simple test. Mini-guts from cystic fibrosis patients don't swell when exposed to cholera toxin, because mutations have crippled the ion channel that the toxin work on and normally force to open.
The ion channel exchange salt and water through the cell membranes and cystic fibrosis patients are protected from the massive diarrhea cholera causes.
The cystic fibrosis drug Orkambi can restore function to the full-length protein if the mutations 'just' cause a misfolding. Thus the mini-guts will swell rapidly if the drug has an effect.
Focusing on these the researchers successfully edit a representative mix of mutations - essentially changing the stop codon and turning the protein back to its original amino acid sequence with an editing efficiency of up to 10%.
Off-target mutations are a concern of the 'classic' CRISPR/Cas9 editing, that relies on double-strand DNA-breaks and homology-dependent repair.
But base editors are deemed much safer because they only nick one DNA-strand and to carefully check for deleterious off-target mutations they use whole-genome sequencing on the edited cells.
»Much to our surprise, we didn't find any off-target mutations. So it's an extremely clean procedure,« says Hans Clevers.
Without the off-target concern, this not only means the method is safe but also that the editing efficiency could be ramped up by merely doing multiple rounds of base editing.
One great thing about organoids is that they are clonal, and everything is done in the lab outside the patient.
»So you can test for any off-target mutations before you transplant back to the patient,« says Hans Clevers.
»I think it's a significant advantage. You can do it hundreds of times and just pick the ones that worked exactly as you hoped. In vivo, you have to live with what comes out of the experiment.«
And another advantage working ex vivo is that circumvent problems with the immune system. The editing components are transferred without using viral vectors, and the Cas9 fusion-protein is long gone by the time the organoids would be transplanted back to the patient and come in contact with the immune cells.
So all in all, the combination of base editors and organoids seem to be an efficient and safe path to a cure for hereditary diseases using gene therapy.
This begs the question of when the first cystic fibrosis patients may benefit from this organoid CRISPR gene therapy? It turns out the answer is more complicated than it might seem.
»If this is going to go into clinical trials, the field first has to show that organoids can be safely transplanted‚« says Hans Clevers.
Currently, there is no clinical practice for transfecting expanded cells whether it be induced pluripotent stem cell (iPS), embryonic or adult stem cells, or organoids. For each organ, the procedure would have to be worked out and optimized. But the first clinical trial using organoids is underway in Tokyo, Japan and maybe a first step for establishing a safe practice for transferring organoids back to patients.
The mini-guts are grown from tiny samples of the tissue lining the gut.
The tissue is incubated in a mix of growth factors to let the gut stem cells replicate. After 2-3 weeks, small organoids about a millimetre in diameter emerge and can be harvested. These organoid lines can grow indefinately. They are stored and managed by the foundation Hubrecht Organoid Technology and University Medical Center Utrecht and can be requested by researchers.
But the new study showing that gene therapy using base editors on human organoids works and is safe raise hope for thousands of other genetic disorders. Organoids can be grown from almost every internal organ - the heart being the noticeable exception - and in principle, the approach can be extended to hereditary diseases in other organs.
They would probably be better suited for a start.
»I guess, cystic fibrosis will not be the disease of choice to start with. Because it's multiple organs that are affected by cystic fibrosis - the lungs but also the pancreas and the liver, gallbladder, and gut. For the first time in patients I would rather go for a liver disease like alpha-1 antitrypsin deficiency, where you only have one organ to transplant,« says Hans Clevers.
The bottom line for cystic fibrosis is that a gene therapy seems feasible, but other hereditary diseases may well get there first.