
Aldevron Joins EGMEDC to Turn Genomic Promise Into...
May. 6, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

For years, Jin-Soo Kim and his team tried and failed to use CRISPR to edit mitochondrial DNA to create mitochondrial disease models and correct disease-causing mutations. It was difficult, if not impossible, to deliver the guide RNA required for targeted CRISPR-Cas9 editing into the membrane-bound mitochondria. The repair of double-stranded breaks in mitochondrialDNA (mtDNA) also presented a major obstacle to successful editing; homology-directed repair and non-homologous end-joining do not occur efficiently in the mitochondria. It seemed like a lost cause.
Then came the breakthrough Kim had been waiting for - a 2020 Nature paper by Professor David Liu’s lab at Harvard, describing the use of interbacterial toxins to base-edit mtDNA in a human cell line. Kim wasted no time adopting the novel method, and in less than one year after its publication, his lab used the technique to successfully edit mtDNA in mice. The results were recently reported in Nature Communications.
»The mitochondria appears to be the last frontier in the field of genome editing. Its genome is small in size, compared to the nuclear genome, but there are many diseases caused by mutations in the mitochondria,«Kim said when asked how he came to be involved in the field of mitochondrial engineering.
In 2013, Kim’s lab published a genome-scale library of TALENs targeted to each human protein-coding gene using a high-throughput Golden Gate cloning system. It was a huge effort, aiming to enable efficient and scalable gene editing in humans. The advent of CRISPR, however, made the TALEN library almost obsolete. Years later, TALENs would help piece together the puzzle of mitochondrial genome engineering.
So how can interbacterial toxins be used to edit mtDNA? The toxin utilized by Kim’s team is known as DddAtox, encoded in the genome of the bacteria Burkholderia cenocepacia and described in the Liu lab’s paper. DddAtox is a cytosine deaminase – an enzyme employed by the immune system of certain bacteria in order to disable other bacteria. When used to attack another bacterial cell, DddAtox catalyzes the hydrolytic removal of cytosine from DNA, replacing it with uracil.
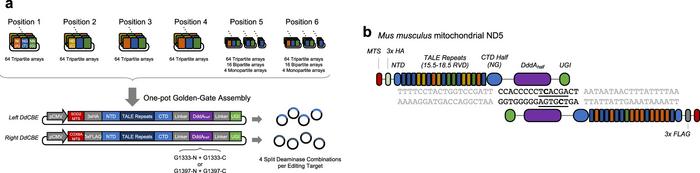
Schematic of the Kim lab’s method for mitochondrial DNA editing using DdCBEs. (A) One-pot Golden-Gate assembly for efficient DdCBE construction and (B) DdCBEs editing the target ND5 gene in mouse mitochondrial DNA. From Nat Comm (2021) 12:1190.
Kim explained how his TALE proteins became suddenly useful again: »Before the CRISPR era, researchers used transcription-activator like effector (TALE) proteins to recognize a pre-determined DNA target site. TALE proteins can be fused to DddAtox, a cytosine deaminase moiety derived from bacteria to edit mitochondrial DNA in a targeted manner,« Jin-Soo Kim said.
Unlike other similar enzymes, DddAtox is unique in that it can generate these changes in double-stranded DNA. By combining DddAtox with custom-designed TALE proteins and a uracil glycosylase inhibitor (UGI), Kim’s team generated what are now known as DddA-derived cytosine base editors (DdCBEs), capable of producing C to T edits in the mitochondrial genome. In order to use DddAtox as a base editor, it must first be split into two, non-toxic halves, which are subsequently brought back together at the target site.
Studies have frequently indicated the lack of accurate models of mitochondrial diseases. Such models are a necessity in order to advance research on new treatments and potential cures. Kim and his team aimed not only to prove that DdCBEs could be used to edit the mitochondria and potentially cure mitochondrial diseases in the future, but that they could also produce precision in vivo models of the diseases.
»Animal models are essential for testing anti-oxidant drug candidates, which is not a cure [for mitochondrial disease] but can be an efficient treatment,« he remarked.
The paper specifically targeted the ND5 gene, which codes for the NADH-ubiquinone oxidoreductase chain 5 protein. The protein is involved in the mitochondrial electron transport chain. Mutations in ND5 are associated with several types of mitochondrial disease, including mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS), Leigh’s syndrome, and Leber’s hereditary optic neuropathy.
Kim’s team sought to generate several different mutations in the ND5 gene; two silent mutations (m.C12539T and m.G12542A), a point mutation which is known to cause mitochondrial diseases (m.G12918A), and a nonsense mutation causing a premature stop codon (m.C12336). Before in vivo experiments in mouse embryos, the team first tested these edits in vitro using the NIH3T3 mouse cell line.
»We know, from our previous experiences with CRISPR systems, ZFNs, and TALENs, editing efficiencies in embryos are higher than in cell lines. Editing efficiencies in the mouse cell lines were good enough for us to go ahead and make animal models,« he explained, adding that his team chose the most active DdCBEs for each edit to proceed with creating animal models.
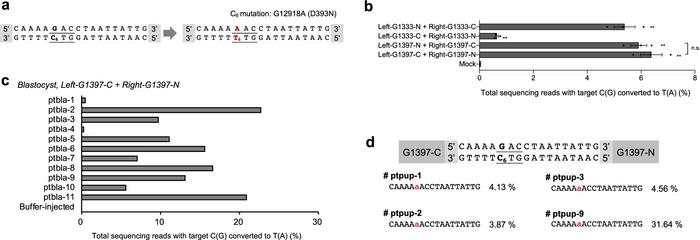
Mouse mitochondrial ND5 G12918A mutation induced by DdCBEs. (A) The DdCBE target for generating the m.G12918A point mutation (codon underlined, locus in red), (B) efficiency of C to T base editing NIH3T3 cells using DdCBEs, (C) base editing efficiency of the m.G12918A point mutation in mouse blastocysts, and (D) ND5 point mutation in F0 mouse pups. From Nat Comm (2021) 12:1190.
Editing efficiencies in cell lines were up to 19% for some mutations, while in embryos, efficiencies were as high as 25%. Successfully edited embryos were subsequently implanted into surrogate mothers to determine if the edits were retained after birth. The majority of the resulting F0 pups retained the edits with efficiencies of up to 31.6% for the disease-causing mutation and 57% for the premature stop codon.
Although the paper states that the disease-causing mutation did not result in an apparent phenotype in F0 pups immediately after birth, it postulates this is most likely due to both the young age of the pups and heteroplasmy. Perhaps surprisingly, the premature stop codon in ND5 was not lethal to the pups. The team also investigated the heritability of the two silent mutations by crossing an edited female F0 mouse with a wild type male. The resulting F1 mice retained the edits with efficiencies of up to 26% across various tissues.
The success of the Kim lab’s in vivo experiments, particularly in generating the m.G12918A point mutation in ND5 which causes mitochondrial disease, is a significant breakthrough for mitochondrial disease research. Creating precision models of mitochondrial disease will no doubt lead to a flood of pre-clinical research testing drug candidates and refining current treatments for these debilitating illnesses.
David Liu shared his thoughts on the rapid progress of Kim’s lab with CRISPR Medicine News: »I was really impressed—blown away, actually—by how quickly Jin-Soo Kim and his talented coworkers achieved the application of these mitochondrial base editors in mouse embryos to create animal models of human mitochondrial genetic diseases,« Liu said.
»I’m hopeful the Kim lab paper will also stimulate additional researchers to explore these tools that allow us for the first time to make precise changes in the sequence of mitochondrial DNA in living cells and animals.«
Regarding the safety of this new method in potential therapeutic applications, Kim stressed the need to ensure there are no off-target effects before testing can begin in human subjects.
»[Potential off-target effects] are a legitimate concern. It is important to develop unbiased methods for profiling genome-wide off-target sites and for reducing off-target effects,« he said
Not only can DddAtox edit nuclear and mitochondrial DNA, it can also edit the chloroplasts of plants, as Kim’s group describe in a recent preprint. This research opens new doors for plant biotechnology, which has previously been hindered due to an inability to engineer plant organelles.
While the success of the method to date is astounding, engineering is not yet possible for every mitochondrial gene. A key limitation of DddAtox is that it only produces certain edits and cannot be used to generate all the possible mutations in mtDNA that cause mitochondrial disease. However, Kim and his team remain hopeful that the technology will progress rapidly and overcome this obstacle.
»The current system allows TC to TT edits. It will be possible to engineer DddAtox or discover its homologs in nature with altered or expanded editing capacity,« he postulated.
When asked to speculate about when clinical trials for mtDNA editing in human subjects could begin, Jin-Soo was quietly optimistic.
»Honestly, I don’t know [how far away clinical trials will be]. In retrospect, CRISPR was fast. It took only several years from technology development to clinical trials. I hope mitochondrial editing is also fast,« Kim concluded.
With the promising results so far, it will certainly be interesting to see what developments occur in mitochondrial engineering research in the coming years.
Link to the original article in Nature Communications: Mitochondrial DNA editing in mice with DddA-TALE fusion deaminases
Rebecca Roberts is a molecular biologist and science writer/communicator based in Queensland, Australia.





