CMN Weekly (26 July 2024) - Your Weekly CRISPR...
CMN Weekly
Jul. 26, 2024


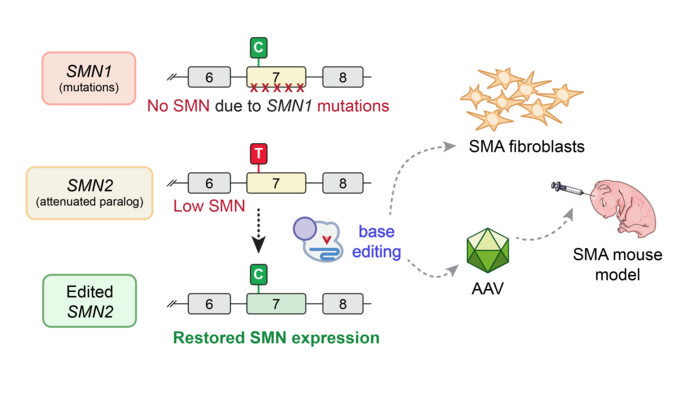
Spinal muscular atrophy (SMA) is the most common cause of infant genetic mortality, occurring in approximately 1 in 10,000 live births. This autosomal recessive disease is caused by loss-of-function mutations in the SMN1 gene, which encodes the survival motor neuron (SMN) protein, a regulatory protein that impacts various aspects of RNA metabolism and which is critical for the maintenance of spinal motor neurons.
Exon 7 deletion is the most common mutation in SMN1, which occurs in more than 90 % of SMA cases, and which abrogates SMN protein function. SMN deficiency eventually leads to degeneration and death of motor neurons, and progressive weakness.
SMA occurs along a continuum of severity, and Type 1 SMA is the most severe and most prevalent form of the disease, affecting approximately 60% of all SMA patients. These patients experience early-onset motor weakness, respiratory failure and typically die by the age of two years.
One factor that governs SMA progression and pathology is the number of copies of SMN2 present in the genome. SMN2 is an SMN1 paralogue, which differs from SMN1 by a single C•G-to-T•A base pair change at position 6 in exon 7 (referred to as C6T). This mutation leads to altered splicing of pre-mRNA and frequent skipping of exon 7 in the SMN2 transcript, producing SMN that is rapidly degraded. Each copy of SMN2 produces about 10% of the amount of full-length SMN protein produced by healthy SMN1, so the more SMN2 copies an SMA patient has, the less severe their disease phenotype typically is.
Three agents are approved by the FDA and the European Medicines Agency for the treatment of SMA. Two of these, Nusinersen (marketed as SPINRAZA® by Biogen) and Risdiplam (marketed as Evrysdi by Genentech) are splicing modifiers that can enhance SMN2 expression by increasing the retention of exon 7 in SMN2 transcripts. Zolgensma (marketed by Novartis) is a single-dose transgene therapy that delivers a functional copy of SMN1 via adeno-associated virus 9 (AAV9), a non-replicating viral vector.
These agents have dramatically improved the prognosis of patients living with this disease, but with limitations. Concerns exist about the long-term efficacy of Zolgensma, while splicing modifiers require repeat dosing. The latter is a particular concern with Nusinersen, which is administered via spinal injection.
To address the urgent need for curative SMA therapies and considering the limitations of SMN1-targeting strategies, separate teams at Massachusetts General Hospital (MGH) and the Broad Institute each devised a novel base-editing approach that would restore SMN protein expression.
In one manuscript, the work was led by Dr. Christiano Alves, a junior faculty member (Instructor) in the laboratory of Dr. Ben Kleinstiver in the Center for Genomic Medicine at MGH, collaborating with Dr. Rashmi Kothary at the University of Ottawa; the other study was led by Dr. Mandana Arbab, a senior fellow in the laboratory of Dr. David Liu at the Broad Insitute.
The base-editing strategies developed in both laboratories sought to return SMN to normal levels regardless of the patient’s SMN1 mutation by editing the C6T mutation in SMN2. The MGH team, whose work is the focus of this feature article, tested a range of different base editors and guide RNAs (gRNAs), and in some cases they observed up to 99% intended editing in SMA patient-derived fibroblasts, which coincided with increased SMN2 expression and SMN protein levels.
“Our publications are the first to demonstrate that gene editing can dramatically change SMN protein levels across different cell lines, including those that have the SMA phenotype.”Dr. Christiano Alves
The findings from the MGH team were recently shared in a manuscript on the pre-print server bioRxiv. Findings from the Broad team were recently published in Science. Together, these studies demonstrate the potential to develop a long-lasting base-editing treatment for SMA that may be preferable to current nucleic acid, small molecule, or gene replacement therapies.
Commenting on the novelty of their approach, Dr. Alves says:
»Our publications are the first to demonstrate that gene editing can dramatically change SMN protein levels across different cell lines, including those that have the SMA phenotype.«
Drs Alves and Kleinstiver chose to work with adenine base editors (see Fact Box) to revert the exon 7 mutation in SMN1 via an A•T-to-G•C base substitution. In this way, the base-edited SMN2 gene should result in sustained increased SMN expression, to compensate for the lack of functional SMN protein in SMA patients. Dr. Alves explains the rationale behind this strategy in more detail:
»C6T is a silent mutation, which means that its translationally innocuous in a coding region of DNA. However, it occurs in a very important spliceosome-binding site. Spliceosomes are responsible for splicing preRNA, which basically means removing introns and assembling exons in the correct order. When this binding site is defective, as in SMN2, exon 7 is not “spliced” into the mRNA transcript and very little full-length, functional SMN is produced. Reverting the C6T mutation in SMN2 should increase retention of exon 7 and thus improve overall expression of full-length SMN protein.«
Importantly, because the team’s strategy only targets the C6T site, the existing SMN2 promoter and canonical endogenous regulatory mechanisms are maintained. This contrasts with Zolgensma, where an exogenous promoter is used to drive SMN1 expression from a transgene, as Dr. Alves points out: »We don’t know precisely how this external promoter is being regulated, but there is some evidence described in an article published in Nature Neuroscience* regarding neuron-specific negative effects of long-term SMN overexpression with an exogenous promoter.«
During an initial screening phase, the MGH team determined which combinations of Cas9 variants, deaminase domains and guide RNAs were most effective at editing the C6T site in HEK 293T cells, a widely used human cell line that is easy to transfect and study.
The researchers faced two challenges when designing the base-editing strategy for C6T. Firstly, because wild-type SpCas9 (Cas9 from Streptococcus pyogenes) has a strict protospacer motif (PAM) requirement (only recognising NGG), the target adenine is at the border of the edit window.
The edit window is a stretch of nucleotides within which a base editor can operate, and SpCas9’s requirement for NGG limits where the Cas9 nickase can be positioned. To get around this issue, which is a bottleneck for base editing with SpCas9 in general, Dr. Kleinstiver’s team and others have developed engineered Cas9 variants that have less restrictive PAM requirements. These so-called PAM-flexible or near-PAMless variants allow more flexibility in positioning the edit window. The second challenge is that the target adenine is next to three other adenines, which increases the potential risk of bystander editing (unintended edits within the edit window).
Taking those challenges into account, the team created a panel of adenine base editor (ABE) constructs using several Cas9 variants (WT SpCas9, the previously developed PAM-flexible variant SpG, the PAMless variant SpRY, and other relaxed PAM variants developed by the Liu lab), three deaminase variants (ABEmax, and 2 newer variants, ABE8.20 and ABE8e, that have been shown to improve on-target editing), and designed seven different gRNAs against sites harbouring various PAMs, tiling the edit window of the ABEs to place the target adenine in positions A4-A10 of the target site.
The team observed the highest levels of on-target editing with minimal bystander editing in HEK 293T cells transfected with combinations of either wild-type Cas9-ABE8e with gRNA A10 or SpRY-ABE8e with gRNA A8. Lower on-target efficiencies were observed with SpG and other relaxed PAM variants.
The team then combined ABE8e-SpRY with different gRNAs that target C6T, ISS-N1 and ISS+100 (ISS-N1 and ISS+100 are sites located in intron 7 that, like C6T, regulate SMN2 splicing). Editing efficiency was similar when multiplexing with two guides compared with a single guide.
Dr Alves notes the potential of such a dual-editing approach to further increase the likelihood of exon 7 inclusion in the SMN2 transcript, and this is something the team will explore further: “If you want to use two different guides to edit two independent sites in DNA, SpRY has the capacity to recognise both sites at the same time. Since every AAV9 transfection is a “insult” to the cell, multiplexing gRNAs with the same base editor would help you to reduce the number of transfections.”
A base editor is a combination of a Cas9 variant called nickase fused with a deaminase. When directed by a guide RNA to genomic sites, base editors can make single base substitutions. Adenine base editors (ABEs) convert A-to-G and cytosine base editors (CBEs) convert C-to-T), but unlike Cas nucleases, base editors are less likely to produce many DSBs. Instead, a base editor nicks the complementary strand, then the cell repair process uses the edited base as a template to repair this nick.
Having identified ABE8e-SpRY and gRNA A8 as the optimal pair to edit C6T in HEK 293T cells, the team then edited SMN2 C6T in fibroblast cells derived from five SMA patients, all of whom harbour a homozygous exon 7 deletion in SMN1. Inclusion of a GFP tag in the plasmid encoding ABE8e-SpRY allowed the researchers to selectively isolate only those fibroblasts that were successfully transfected for further analysis.
Targeted sequencing of GFP+ SMA fibroblasts revealed >90% editing of the C6T site in three of the five cell lines compared to control cells, with around 60% editing of the C6T site for the other two lines, and minimal (<1%) bystander editing of adjacent adenines for all cell lines.
When investigating the impact of C6T editing on SMA phenotype in the three highly-edited lines, the team found that expression of full-length SMN2 mRNA (i.e. that included exon 7) was six-fold higher than in unedited fibroblasts as determined using droplet digital PCR, while SMN protein levels were 3.5 to 15 times higher in edited fibroblasts compared to control cells, when assessed by immunoblot or ELISA, respectively.
Having succeeded in editing C6T in HEK 293T cells and SMA-fibroblasts with concomitant increases in SMN2 and SMN expression, the team assessed the specificity of their approach in various ways.
Firstly, in silico off-target predictions using CasOFFinder nominated between 11 and 50 putative off-targets for wild-type SpCas9, SpRY or SpRY-HF1 (a SpRY variant that harbours fidelity-enhancing mutations and which has been shown to almost completely eliminate off-target edits in cells), in combination with the best-performing C6T-targeting gRNAs (A5, A7, A8 and A10).
The in vitro assay CHANGE-seq was then used to nominate putative off-target sites. In CHANGE-seq, genomic DNA from SMA-fibroblasts is treated with the Cas9-gRNA complex and then on- and off-target cleavage sites are identified via sequencing. The team tested various combinations of gRNAs A5, A7, A8 and A10 with either wild-type SpCas9, SpRY or SpRY-HF1. Overall, these experiments revealed that editing with SpRY was associated with a higher level of off-targets compared to wild-type Cas9 or SpRY-HF1.
Finally, to assess potential off-target editing in cells, the team performed targeting sequencing of the top 34 CHANGE-seq detected off-target sites using genomic DNA isolated from the five SMA fibroblast cell lines that were edited with ABE8e-SpRY and gRNA A8. This revealed specific editing in three of the SMA cell lines, with only two adenines in two off-target sites demonstrating higher than background editing.
In contrast, a similar analysis in HEK 293T cells edited with ABE8e-SpRY and gRNA A8 revealed off-target editing in at least one adenine in 29 of the top 34 CHANGE-seq detected off-target sites. The team suggests that the different outcomes seen in edited SMA-fibroblasts compared to HEK 293T cells may be attributable to differences in transfection efficiencies and expression levels of the two cell types, though this merits further investigation.
Commenting generally on the off-target analyses, Dr. Alves says: »With PAMless Cas9 variants, if you can target any site, you can in many cases observe an increase in off-target editing. SpRY is a great research tool because you’re not limited by the PAM, but for therapeutic use, it can be beneficial to use a PAM-specific enzyme or high fidelity variants that reduce the risk of off-target edits.«
Having optimised the C6T base-editing strategy in an easily tractable workflow based on cell lines, the team wanted to investigate the efficacy of in vivo SMN2 editing in SMNΔ7 mice, which express human SMN2 as a transgene, and which are used as a model for severe SMA.
Following pilot experiments to assess delivery and administration in HEK 293T cells and neonatal P1 mice, respectively, the team chose to use AAV9 as the transduction vector for in vivo delivery of the base-editing reagents. Given AAV9’s relatively small payload capacity, the team opted for a dual-vector delivery system, whereby ABE8e-SpRY and gRNA A8 were split between two separate AAV9 vectors that were injected directly into cerebrospinal fluid via intracerebroventricular injection.
Sequence analysis revealed approximately 5% on-target C6T editing in the tissues of primary interest, i.e., the spinal cord and brain. Notable C6T editing was also observed in the liver, heart, skeletal muscle, with low levels of bystander editing.
Given the disease severity and short life span of the SMNΔ7 mouse, which typically doesn’t survive for more than 15 days, it was not possible to assess SMA phenotype following in vivo SMN2 editing, but this is something the team will address soon, using an alternative SMA mouse model with a less severe phenotype that will also permit long-term monitoring of the effects of base editing SMN2.
Dr. Alves points out that the timing of base editing may also be an issue: »We treated neonatal mice, but phenotypic rescue may require in utero treatment, before there is irreversible pathology.«
In addition, Dr. Alves comments: »Another approach would be a combination therapy with other SMA drugs to extend the survival in this mouse model as demonstrated in the recent Liu’s lab publication in Science.«
»The combined results from our studies highlight the potential of base editors to effectively and safely upregulate SMN expression to overcome SMA-related pathologies«, said Dr. Kleinstiver. »We are excited about the implications of this work for SMA and other diseases.«
As well as following up on the outcomes of in vivo SMN2 base editing, the Kleinstiver lab is also working on a couple of other potentially game changing refinements in base editors, including deaminases with a shorter edit window, which would reduce bystander edits, and smaller versions of Cas9 proteins that would enable the delivery of the entire base editor construct in a single vector. Using one instead of two AAV9 vectors would be a welcome approach to reduce viral dosing and the side effects associated with viral vectors.
Dr. Alves closed the interview with a final thought: »Motor neurons are primarily affected when you don't have SMN, but many other tissues may be affected as well. Nusinersen, which is so far the most-commonly used SMA treatment, only target the CNS. In my opinion, there needs in the SMA community to further understand the effects of SMN replacement in the entire body«.
Link to original manuscript on bioRxiv
Base editing as a genetic treatment for spinal muscular atrophy
* Van Alstyne M, Tattoli I, Delestrée N, Recinos Y, Workman E, Shihabuddin LS, Zhang C, Mentis GZ, Pellizzoni L. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat Neurosci. 2021 Jul;24(7):930-940. doi: 10.1038/s41593-021-00827-3.
Karl Torbey MD writes about science and medicine and is based in the United States.
To get more of the CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.
ArticleInterviewNewsAdeno-associated virus (AAV)Spinal Muscular Atrophy, SMARare DiseaseBase editors