CRISPR-Armed Phages Clear First Human Safety Bar
CMN Interview
Mar. 3, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Most people will have heard stories of elite athletes dying suddenly on the basketball court or playing field, despite being in peak physical condition and often with no prior symptoms.
»These cases are almost always due to hypertrophic cardiomyopathy,« says Professor Eric Olson, chair of department of molecular biology at the University of Texas Southwestern Medical Centre.
Hypertrophic cardiomyopathy (HCM), the most prevalent genetic heart disease, affects more than 0.5% of people in the US. Thickening of the heart muscle associated with HCM causes arrhythmia, heart failure, and sudden cardiac death. The condition often goes undiagnosed, and heart transplant is the only known cure.
Olson’s latest work attempts to level the playing field by employing base editing to correct one of the mutations that causes HCM. Published in Nature Medicine, the proof-of-concept study demonstrates that with a single nucleotide substitution, a subset of HCM patients could potentially be cured of the disease.
As Olson remarks, the study is a promising first step toward creating a novel in vivo gene therapy for HCM:
»[In this paper] we’ve shown that we can effectively deliver a split base-editing system to the heart, we can correct the target mutation with minimal off-target editing, and it can confer therapeutic benefit.«
“...heart cells turn over very slowly, if at all: the heart cells that are beating in you and me have been beating forever. So, once you correct the mutation, it should be a one-and-done therapy. It's not a tissue like the liver that's undergoing constant cellular turnover”Eric Olson
A developmental and molecular biologist, Olson originally studied the mechanisms by which genes can be switched on and off during development. He has since dedicated his career to understanding and developing treatments for a variety of heart and skeletal muscle disorders.
When you’ve spent enough time working on muscle diseases, Olson says, you will ultimately encounter what he calls the ‘Mount Everest of muscular disorders’: Duchenne muscular dystrophy (DMD).
»Muscle and heart are plagued by devastating diseases that destroy the quality of life, and life itself. And many of those diseases have been refractory to any type of therapies. DMD is the most devastating of these muscle diseases, and it has defied every treatment,« Olson comments.
Like so many other researchers working in the field of genetic disease, Olson was excited by the advent of CRISPR gene-editing technology, primarily because of his work on DMD.
»It offered the opportunity to go after the cause of the disease – namely, the genetic mutation – rather than the symptoms of the disease,« he recalls.
Olson and his team built on their extensive work on DMD and expanded into potential therapies for HCM. In this latest study, their target was a mutation in the myosin heavy chain 7 (MYH7) gene. MYH7 codes for β-myosin heavy chain, a motor ATPase that incorporates into cardiac muscle filaments and affects their contractions.
The pathogenic G to A mutation in MYH7 (c.1208G>A; p.R403Q) causes severe, early-onset HCM; around half of the patients who carry this mutation die by age 40. Fortunately, this mutation can be targeted by base editors.
Base editor (BE) systems are derivatives of the original CRISPR editing system which fuse either a Cas9 nickase (nCas9) or a catalytically-dead Cas9 (dCas9) to deaminase enzymes to induce single nucleotide substitutions in DNA. Often hailed as a safer alternative to CRISPR-Cas9 editing, BEs do not induce double-stranded breaks in the genome.
To correct the G to A mutation, Olson and his team required an adenine base-editing (ABE) system, which uses a bacterial deoxyadenosine deaminase to catalyse the substitution of A/T base pairs for G/C base pairs.
After generating induced pluripotent stem cells (iPSCs) from healthy donors, Olson and his colleagues inserted the pathogenic mutation into the MYH7 gene using CRISPR-Cas9-based editing. They then used these cells to compare different ABEs paired with a range of Cas9 variants and using a range of different sgRNAs.
»We wanted to identify the optimum guide and the optimum gene-editing strategy in the human genome, so we screened several different types of base editors and guide RNAs in order to find the optimum combination [to correct the mutation],« Olson explains.
Based on this process, the team chose the ABEmax-VRQR system, made up of ABEmax, which has a narrow editing window, fused to SpCas9-VRQR, a Cas9 variant with very stringent protospacer adjacent motif (PAM) requirements. The team subsequently used the system to correct the MYH7 mutation in HCM patient-derived iPSCs, achieving extremely high editing efficiencies of up to 99%, with little to no off-target or bystander editing.
Next came the analysis of cell function post-editing, for which the group differentiated the edited and non-edited HCM iPSCs, as well as healthy donor iPSCs, into cardiomyocytes. HCM cardiomyocytes typically display increased force generation and ATP consumption compared to healthy cardiomyocytes. Olson and his colleagues were able to determine that the corrected HCM cells had reduced hypercontractile force generation and energy consumption compared to non-edited HCM cells.
The next step was to prove that the system could work in vivo. For this purpose, Olson and his team used a humanised mouse model. While the amino acid sequence of the relevant protein is 100% identical in mouse and human around the mutation, the DNA sequence is significantly different, so they replaced part of the mouse gene with the human MYH7 gene carrying the pathogenic mutation.
Despite the challenges associated with in vivo gene editing, Olson asserts that the heart is an attractive therapeutic target. The heart is a consolidated tissue, meaning that the gene-editing components don’t have to be delivered throughout the body. There are also adeno-associated virus (AAV) serotypes that display very strong tropism for the heart, allowing viral doses to be lowered and therefore making it a safer delivery strategy.
»Finally, heart cells turn over very slowly, if at all: the heart cells that are beating in you and me have been beating forever. So, once you correct the mutation, it should be a one-and-done therapy. It's not a tissue like the liver that's undergoing constant cellular turnover,« Olson continues.
Owing to the size of the editing components, the group split the BE system, packaging them into two AAV9 vectors along with a heart-specific promoter and the best sgRNA. Trans-splicing inteins were employed to reconstitute the BE system once the proteins were expressed.
Olson was cautiously optimistic about testing the system in vivo, but the results were even better than expected. After the system was delivered to diseased mouse pups, subsequent analyses revealed that editing efficiencies were above 30%, and the correction was able to prevent onset of HCM, with edited mice showing similar cardiac dimensions to healthy control animals (Figure 1).
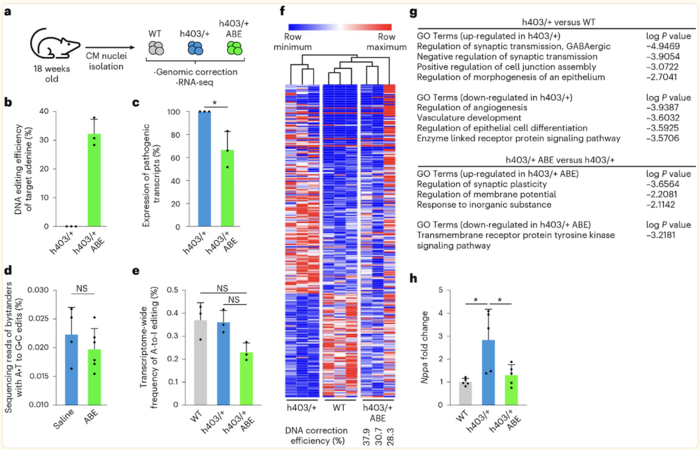
The results of the study are very promising, and Olson’s team continues to validate their strategy. The next step is to generate more data in a large animal model – in this case, a pig, which is the preferred option in the field of cardiology. They are also intent on making this potential therapy as safe as possible, and are currently investigating improved delivery strategies, such as lipid nanoparticles (LNPs), in an effort to move away from viral delivery.
»Something we are also working on, which is really important, is getting rid of Cas9 after editing is complete. You don't want a high level of Cas9 in human cells long-term. If you could eliminate it, that would be advantageous. These are some of the issues that will need to be confronted in the future,« Olson adds.
As the MYH7 base-editing therapy moves toward clinical trials, Olson and his team are also testing editing strategies for other HCM-causing mutations. They have also recently published key papers demonstrating the correction of pathogenic mutations in RBM20, which cause dilated cardiomyopathy (DCM), and modification of a signal-responsive amino acid in CaMKIIδ, which causes various cardiac disorders.
It's estimated that one in every 250 people has a genetic mutation that leads to heart diseases such as HCM and DCM, but the team is determined to conquer as many of these as possible, as Olson concludes:
»These mutations are scattered throughout the different genes that are involved in cardiac function, which means you can’t develop a one-and-done consolidated editing strategy that can just fix everything. So there are a lot of really interesting challenges ahead, but I'm super excited about it. We’ll confront those challenges one at a time.«
Link to the original article in Nature Medicine:
Base editing correction of hypertrophic cardiomyopathy in human cardiomyocytes and humanized mice
Rebecca Roberts is a molecular biologist and science writer/communicator based in Queensland, Australia.
To get more of the CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.