
Aldevron Joins EGMEDC to Turn Genomic Promise Into...
May. 6, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Caribou Biosciences' pipeline boasts three CAR-T cell therapeutic candidates for haematological cancers, two of which are being evaluated in clinical trials, one CAR-NK cell candidate for solid tumours in pre-clinical development, and two undisclosed discovery CAR-T cell programmes in collaboration with AbbVie.
The company’s front-runner candidate CB-010 was the first allogeneic CAR-T cell therapy with a CRISPR-mediated PD-1 deletion to be cleared for a clinical trial (in 2020), and it is currently being evaluated in the Phase 1 ANTLER trial for relapsed or refractory non-Hodgkin lymphoma.
Clinical data from the ANTLER trial released in 2022 revealed that CB-010 was the first allogeneic CAR-T cell therapy to achieve 100% complete response rate at the first dose level, which means that no cancer was detected in the first six patients treated with CB-010 as their best response. Additional clinical data will be forthcoming before the end of 2023.
CRISPR Medicine News recently spoke with Caribou’s Chief Scientific Officer Steve Kanner, Ph.D., to learn more about the company’s unique approach to genome editing, and how it is leveraging its platform to develop novel off the shelf treatments for a range of cancers.
- Hi Steve, thanks for speaking with us. I understand that Caribou’s Cas12a chRDNA technology takes a unique approach to CRISPR-Cas editing. Can you tell us why Caribou has developed its own version of CRISPR-Cas?
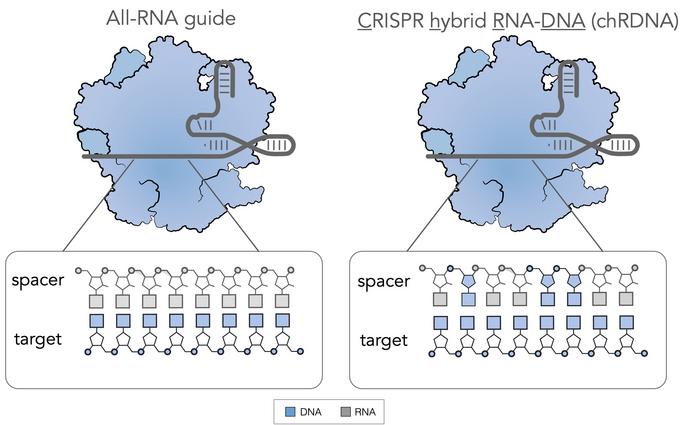
The original CRISPR technology involved Cas9 plus a dual RNA guide composed solely of RNA. There are two components to these guides. One component is called the CRISPR RNA (crRNA), which interacts with the chromosomal DNA and helps to guide the Cas to the desired site. The second component called a tracrRNA binds to the crRNA through molecular interactions to create a hybrid, which is a stem-like structure that interacts with the Cas and stabilises it.
The process of discovery has led to the development of single guide RNAs (sgRNAs), where both of the above components are present in a single piece of RNA that is easier to manipulate. These all-RNA guides are widely used in first-generation CRISPR-Cas.
One of the things that can happen when using all-RNA guides is that one may observe off-targets when genome editing is performed in human cells. This happens because mismatches between the guide and the chromosomal DNA can still lead to binding, especially if it's an all-RNA guide, because the interaction between an all-RNA guide and a DNA strand is pretty high-affinity.
Off-targets are a major concern during genome-editing therapy development, since they can lead to chromosomal translocations, which could inactivate tumour suppressor genes or activate oncogenes leading to cancer. Caribou set out to address this issue by developing the chRDNA technology. By using a chRDNA (pronounced Chardonnay) instead of an all-RNA guide, we can eliminate or reduce off-target editing as much as possible while still maintaining the necessary level of on-target editing.
- So how does the chRDNA system solve the off-target problem?
chRDNA started when our scientific team discovered that putting DNA into the backbone of an RNA guide led to a detuning of the affinity when there's a mismatch between the guide RNA and a region of genomic DNA. When there is a perfect match, the affinity is sufficient to bring about a conformational change in the Cas enzyme to generate the desired double-stranded break (DSB). But if there's a mismatch, e.g., at an off-target site, the chRDNA detunes the affinity sufficiently to prevent enzymatic reconfiguration so that no DSB occurs.
In practice, we design and develop each chRDNA following identification of strategic locations within each guide that have to be identified empirically, for the most part. The chRDNAs result in the same level of on-target editing as the equivalent all-RNA guide but without detectable off-targets. For any edits, we don’t observe off-targets in the detection assays that we use, where the limit of detection is 1/1000 reads.
- Is it necessary to understand the potential off-target profile for each chRDNA in order to optimise the design, and does this slow down the development process?
Any amount of off-target analysis takes time. To help drive that analysis internally, we developed an unbiased off-target detection assay called the SITE-Seq® assay, which is different from many of the other off-target assays in use. To perform the assay, we isolate genomic DNA (gDNA) from unedited cells, let's say primary T cells from blood. Then we put that gDNA into a test tube together with the Cas of interest and the guide. We can then compare the editing outcome with an all-RNA guide versus a chRDNA guide, and see what the difference is in terms of the DSB pattern in an unbiased and completely biochemical assay. This data provides a map of where to look genomically for potential off-target editing when performing a cell-based validation study.
Developing a chRDNA guide is an iterative process. To preserve on-target and minimise off-target activity, we need to identify any sites within the guide that cannot be changed from RNA to DNA without eliminating Cas activity. We’ve learned a lot through experience and now have the institutional knowledge of where not to add DNA, but there is no predictive tool to design an effective chRDNA. However, when we identify a really good chRDNA, we can use it in multiple programmes.
- I’ve noticed that Caribou’s first candidate CB-010, which is now in a Phase 1 clinical trial, was developed using Cas9, but Cas12a is used in the other pipeline candidates. What is the reason for this switch?
So, Cas12a generates a staggered DSB while Cas9 makes a blunt-ended DSB. We find that the staggered break works better when trying to drive the cell toward homology-directed repair to rejoin the chromosomal fragments after insertion of a desired template. A blunt DSB typically favours the non-homologous end joining machinery, which essentially bridges the chromosomal fragments back together and appears less efficient in terms of driving an insert into the desired DSB site.
On a related note, in both cases (whether we use Cas9 or Cas12a), we infect T cells with adeno-associated virus 6 (AAV6) that has been engineered to harbour the desired gene for insertion. T cells express a receptor for AAV6, which allows us to deliver the DNA template into the cell through viral infection. This results in an abundance of plasmid inside the cell that has homology to the cut site where we’re trying to insert a transgene. This could be a chimeric antigen receptor (CAR) with homology on either side that matches exactly where we generated the DSB in the genomic DNA via electroporation of the CRISPR machinery. The cell’s DNA repair machinery will try to force insertion of the CAR at that site, and this seems to work more efficiently with Cas12a.
With the switch to Cas12a plus the chRDNA approach, we are observing up to 80% insertion rates of our desired inserts, and we have observed ~60% of a population of T cells harbouring double gene inserts that require four total edits (two knockouts and two knock-ins).
- Many new Cas enzymes have been discovered and characterised in the past few years, some are highly compact, while others are reported to be more specific, or more flexible in terms of PAM specificity. Is Cas12a the end of the line for Caribou or are you pursuing other enzymes that might be even better?
I think that all of these different enzymes seem to have a purpose. As one example, some scientists have inactivated the nuclease activity of the Cas to make an enzymatically-dead form (the so-called dCas9), which can then be linked to another enzyme, for instance a transcriptional activator. While alternative gene-editing nucleases are really fascinating from an academic point of view, their therapeutic application is more limited. They could be really good for creating a knockout or making a base change, but not knock-ins or introducing large genes, because they can only introduce small changes, typically a small number of nucleotides.
We’ve performed some comparisons and found that even with an ‘improved’ enzyme that exhibits fewer off-target edits, but using an all-RNA guide, it's not as good as the chRDNA approach with a natural Cas enzyme. So for our purposes, where we are exclusively focusing on ex vivo cell therapies for the moment, we see no better platform than chRDNA. Because we are multiplexing our cell therapies, we need very high on-target editing efficiencies to ensure we have enough therapeutically active cells at the end of the editing process. We can achieve this with the chRDNA technology.
- All of Caribou’s disclosed programmes are within oncology. How did this become the major focus area?
We got into oncology because when the first CAR-T therapies were being approved in 2017, we saw the potential to use our editing technology to introduce some interesting genome edits to healthy donor cells to make newer versions of CAR-T cell treatments that were ‘off the shelf’. For example, we introduced edits that were distinguished in the field that could prevent host rejection during treatment and prolong CAR-T cell activity, or that were resistant to exhaustion through certain checkpoint pathways.
We can see now from our ongoing ANTLER trial that our innovative allogeneic CAR-T cell therapies could be strong competitors to the autologous CAR-T approach that has known drawbacks.
- Can you elaborate on Caribou’s innovative approach to allogeneic CAR-T cell therapies?
We are using armouring strategies to enhance various abilities of allogeneic CAR-T cells. Some edits were previously known in the CAR-T field, but we were the first to implement these in an allogeneic setting.
One of the first edits we implemented was PD-1 disruption. It’s become clear over the last decade that the interaction between the T cell checkpoint protein PD-1 and its ligand PD-L1 prevents T cells from exerting anti-tumour activity. Knocking out PD-1 in CAR-T cells prevents this interaction, and we knew from lymphoma that PD-1 was an important component of anti-tumour immunity.
In pre-clinical studies, we found that mice with a high tumour burden treated with anti-CD19 CAR-T cells in which PD-1 was disrupted kept on living, while mice treated with anti-CD19 CAR-T cells without PD-1 disruption experience tumour recurrence and eventually expired. The difference was like night and day, and we expect to publish those results shortly. PD-1 knockout likely maintains the CAR-T cells in a highly active state during the first few weeks after treatment, which allows them to drive anti-tumour activity before the immune system eventually catches up and wipes them out through other pathways.
In our other CAR-T cell programmes, we’ve also implemented an editing strategy to prolong the longevity of allogeneic CAR-T cells in circulation. Here, we start by knocking out the B2M gene, which results in the loss of all major histocompatibility complex (MHC) class I antigens on the CAR-T cells. This makes the cells invisible to cytotoxic (CD8) T cells. However, removal of all MHC class 1 antigens will actually derepress natural NK cells. To get around this, in our Phase 1 CB-011 programme for relapsed or refractory multiple myeloma, we have site-specifically inserted a gene encoding a B2M—HLA-E-peptide fusion into the T cell genome. We’ve shown pre-clinically that this prevents NK cells and CD8 cells from destroying the CAR-T cells. We hope that this strategy will prolong the longevity of the CAR-T cells in the bloodstream and give them more time to attack tumour cells.
In all of our T cell candidates, we site-specifically insert a proprietary CAR into the T cell genome, while simultaneously knocking out the endogenous T cell receptor gene TRAC gene to remove the native T cell receptor.
- Does Caribou have plans to expand to other disease areas at any point?
While we are focusing on oncology at this time, we are interested in other applications of CAR-T cell therapies beyond cancer. As an example of an alternative application, a couple of articles have been published recently that describe the use of the same CAR-T cells that are used to treat B cell cancers and lymphoma to treat lupus. Lupus occurs when B cells produce anti-DNA antibodies, and lupus patients are often treated with chemotherapy. It was found that lymphodepletion in lupus patients followed by CAR-T cell therapy led to improvements in disease outcome. The explanation is that the diseased B cells that cause lupus were actually depleted and then replaced by healthy B cells when the bone marrow replenished itself after lymphodepletion and CAR-T cell treatment. That’s a fascinating example of how what we are doing at Caribou could be applicable to other disease areas.
We also have a platform where we edit induced pluripotent stem cells (iPSCs) and then differentiate and expand single cell clones into NK cells for oncology applications, for example our CB-020 discovery programme for solid tumours. But a similar approach could also be applied to generate an unlimited supply of many types of cells to regenerate diseased tissue, with genome edits that are relevant to any given disease.
- Thanks a lot, Steve. We look forward to future updates from Caribou Biosciences!
Link to Nature Methods publication describing SITE-Seq off-target detection assay.
Mapping the genomic landscape of CRISPR–Cas9 cleavage.
To view Caribou Biosciences’ complete pipeline, see here.
To get more of the CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.
ArticleInterviewNewsCancerNon-Hodgkin Lymphoma, NHLCancerCAR-TCRISPR-CasCas12Caribou Biosciences, Inc.





