FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

It is essential to quantify the outcome of CRISPR editing experiments to evaluate unintended events, especially for clinical use of the technology. But existing tools are not sufficiently sensitive to separate signal from noise in experiments with low editing rates, and thus low levels of true off-target activity can get lost under the noise. More importantly, these tools cannot readily detect translocations without the need for specially designed cumbersome experiments.
“We have developed a new software tool that analyses NGS data obtained by multiplex PCR from matched treatment and control CRISPR-Cas9 experiments. The tool applies statistical modelling to determine and quantify editing activity”Ayal Hendel
The new software tool, CRISPECTOR, stands out by applying statistical modelling to determine and quantify editing activity. It learns to recognise the specific patterns of background noise coming from next-generation sequencing (NGS) reading errors and from insertion of incorrect nucleotides during the multiplex PCR reactions. When the background noise can be recognised, the signal - i.e., the true editing events - can be detected and quantified much more precisely than by simple subtraction of the control from the treatment, a method other tools have used.
»We have developed a new software tool that analyses NGS data obtained by multiplex PCR from matched treatment and control CRISPR-Cas9 experiments. The tool applies statistical modelling to determine and quantify editing activity,« says Ayal Hendel, who is co-senior author of a paper in Nature Communications where CRISPECTOR was presented last month. Hendel is a molecular biologist at the Bar-Ilan University in Israel. His group has led the work on the biological experiments used to generate data for the development of the software tool.
Zohar Yakhini, a computer scientist and the paper's other co-senior author, spearheaded data analysis and computer programming with his research group at IDC Herzliya and Technion, Israel.
Yakhini explains the software’s statistical approach to off-target detection:
»CRISPECTOR counts and classifies misaligned reads in a CRISPR-edited treatment and an unedited control. Under the assumption that no edits have taken place, we would expect the same proportion of misaligned reads in the two samples. When we observe significantly more misalignments of any given type in the treated sample, we declare the corresponding reads to represent editing events.«
Ayal Hendel will talk about CRISPECTOR at our free CMN Webinar CRISPR Off-Targets that takes place on Wednesday, 23 June.
The CMN Webinars are hosted and organised by CRISPR Medicine News and will give you the latest updates from the CRISPR Medicine field. In collaboration with our growing global CRISPR Medicine community, we will select relevant topics and invite the speakers from the field.
Sign up to the CMN newsletter for announcements about upcoming webinars, and watch past webinars on-demand* here.
*On-demand access is only available for CMN+ subscribers.
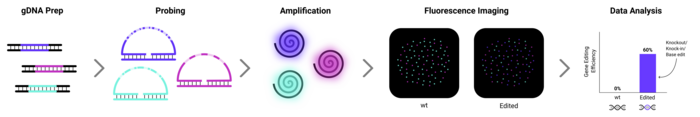
To use CRISPECTOR, a scientist will, in the first place, need a CRISPR-edited sample (the treatment) and untreated control. The scientist must then identify potential off-target sites based on computed or experimental data and select sets of PCR primer pairs to test these sites. Subsequently, each of the two samples is subjected to multiplex PCR, and the amplicons are sequenced by NGS. CRISPECTOR then comes into play by analysing the sequencing data that can comprise many millions of reads.
“To give an example, the probability of being blond is higher for Norwegians than for Israelians. But if I actually observe a blond person in Israel, I can’t readily assume that this person is Norwegian”Zohar Yakhini
»It is essential to understand that CRISPECTOR is not a prediction tool but rather analyses real experimental data,« Yakhini points out and explains:
»We use machine learning for the software’s so-called Bayes classification algorithms that classify sequencing reads as to whether or not they are attesting to an editing event. To give an example, the probability of being blond is higher for Norwegians than for Israelis. But if I actually observe a blond person in Israel, I can't readily assume that this person is Norwegian. I must consider that it is much less probable to encounter a Norwegian than an Israeli in Israel. Bayes classifiers consider these probabilities, called posteriors, and we use them to distinguish signal from noise.«
Using a robust statistical approach, CRISPECTOR can calculate the true editing activity even when error and editing rates are nearly identical. In experimental settings with high error rates and low editing activity, it was also demonstrated that CRISPECTOR beats the competition in detecting off-target editing activity. This comparison was made by feeding the same multiplex PCR NGS data into the new tool and two established ones, CRISPResso2 and ampliCan. The sequencing data was obtained from CRISPR-Cas9 experiments with five different gRNAs targeting five respective genomic loci and covering 226 off-target sites.
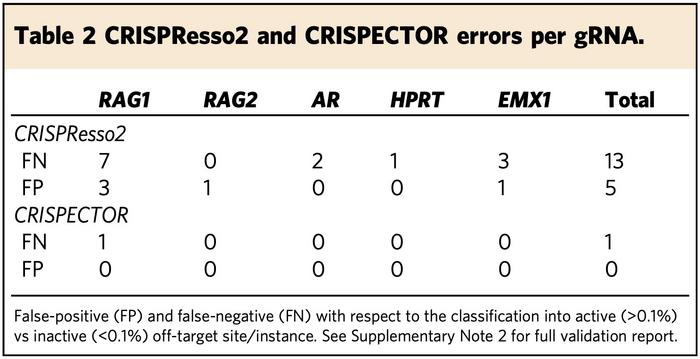
In one experiment, CRISPECTOR and CRISPResso2 disagreed in determining whether 19 instances were edited or not. The researchers then validated these instances through careful human inspection as well as by other means including titration experiments. It turned out that CRISPECTOR called a single false negative, while CRISPResso2 called 13 false negatives and five false positives. Within these same 19 instances, ampliCan called 11 false negatives and two false positives.
One experiment showed that a high rate of 1bp-deletions at the expected cut-site masks the real editing signal in tools using only subtraction. In this case, CRISPECTOR overcame the strong background noise and estimated an editing activity of 0.113%. CRISPResso2 and ampliCan, on the other hand, estimated much lower signals at 0.068% and 0.07%, respectively.
“We thought to ourselves, let's try to see if we can find this data and use it to detect the translocations”Ayal Hendel
However, the real beauty of CRISPECTOR is that its accuracy allows for the detection of translocations from NGS data generated by multiplex PCR. Translocations can arise when two double-stranded breaks on two different chromosomes are repaired by joining the two open chromosome arms to each other. In that case, a primer from one PCR primer pair and another primer from a different pair might pick up the translocation and amplify the sequences flanking the junction.
»This data will be in the files coming from the sequencing experiments, but people are not analysing it. They just put it in the waste bin because the sequences don't map well. We thought to ourselves, let's try to see if we can find this data and use it to detect the translocations,« says Hendel.
Other tools have not used this data before, Hendel explains, because the tools were originally developed for singleplex PCR experiments. When multiplex PCR arrived, the researchers only used the new technology to make several PCR reactions at once. They didn't think that primers from different pairs could work holistically and amplify translocations.
In CRISPR-Cas9 experiments with the different gRNAs, CRISPECTOR found evidence of up to 20 unique translocations that were confirmed by droplet digital PCR using specially designed primers. The translocations originate from the joining of double-stranded breaks at either two off-target sites or one on-target and one off-target site. Translocations were detected at rates ranging from 0.013% to 0.35% of reads.
A software tool like CRISPECTOR might be simple to use - it will give you the result in a few minutes - but it is based on highly complex mathematics, statistics, algorithms and machine learning. We are seeing many such tools used in modern high-throughput biology, Yakhini says:
»Modern biology is becoming more and more data science. In an experiment like our treatment vs. control, you will get tens of millions of reads, and you can’t analyse that in Excel anymore. You have to be aware of what you are doing and use statistics. It is the same in personalised medicine, high-throughput screens etc., where you need to extract a signal. This is data science.«
The two Israeli researchers are now working on improving and expanding CRISPECTOR. The goal is to turn it into an end-to-end solution that will integrate the nomination of putative off-target sites, the design of PCR primers for a focused assay and the analysis of NGS sequencing data.
Academic researchers can use CRISPECTOR for free for non-commercial purposes by accessing the code at http://bioconda.github.io/recipes/crispector/README.html.
Ayal Hendel will talk about CRISPECTOR at our free CMN Webinar CRISPR Off-Targets that takes place on Wednesday, 23 June.
Link to the original article in Nature Communications:
ArticleInterviewNewsNext gen sequencingOff-targetCRISPR-CasCas9





