CMN Weekly (20 February 2026) - Your Weekly CRISPR...
CMN Weekly
Feb. 20, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Homology-directed repair (HDR) has long been the elusive gold standard for precision genome editing. While CRISPR-Cas9 technologies are widely used to disrupt genes via error-prone repair pathways such as non-homologous end joining (NHEJ), introducing precise edits through HDR remains inefficient in most human cells.
To tip the balance toward HDR, researchers have turned to pharmacological inhibitors of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) – a key enabler of NHEJ. Among these, AZD7648 has recently emerged as a front-runner, promising to enhance HDR efficiencies without the toxicity issues associated with earlier compounds.
In his lab at ETH Zürich, Professor Jacob Corn decided to test AZD7648 in genome editing, and initially, he was very encouraged. The compound dramatically increased HDR efficiencies, particularly in primary cells, and importantly, it was well tolerated by the cells. But at a closer inspection, the results seemed almost too good to be true.
»At every locus we tested, the HDR efficiency was remarkably high. In some cases, literally every single allele was HDR, and we didn’t get any indels at all. And so several people in the lab, myself included, thought, this looks great, but maybe we should double check it,« Jacob Corn recalls.
So, the team decided to investigate whether these surprisingly good results might have an alternative explanation. To understand what was really going on, they turned to basic math.
Basic math exposes the hidden flaw
The seemingly flawless results were typically interpreted by dividing the number of HDR events – the numerator – by the total number of edited alleles – the denominator. If the proportion of HDR goes up, it might be because there are more HDR events. But as Corn points out, it could also be because the denominator goes down.
“When we used assays that looked exactly at the site of the cut, then we could see lots of HDR alleles and nothing else”Jacob Corn
To test that hypothesis, the team needed someone who understood what "going down" might actually look like at the DNA level. This is where Grégoire Cullot came in, a postdoc in Corn's lab and the first author of the Nature Biotechnology paper in which the team's results were published.
Cullot had spent his PhD investigating how even a clean cut with Cas9 could sometimes result in unintended large-scale deletions. With that background, he was the ideal person to scrutinise the unexpected aspects of AZD7648's impressive editing performance.
Together with his colleagues, he set out to determine whether the unusually clean editing outcomes were the result of genuine repair or whether something was being missed. To do so, the team combined a suite of complementary techniques, including short- and long-read sequencing, digital droplet PCR, and reporter assays. This multi-pronged approach would allow them to not only confirm the observed HDR rates but also detect any edits that conventional methods might overlook.
Short-read sequencing, widely used to assess genome-editing outcomes, initially confirmed the effect: HDR levels were high, and indels all but disappeared. But short reads only cover a narrow window around the cut site – typically a few hundred base pairs – and therefore miss larger structural alterations. Long-read sequencing, in contrast, spans several kilobases and can expose deletions that stretch far beyond the reach of short reads. This distinction proved crucial.
AZD7648 blows the candles out
In one set of experiments, the researchers edited K-562 and RPE-1 p53−/− cells with and without AZD7648 and compared the results. As shown in Figure 1b, short-read sequencing revealed a striking dominance of HDR (blue bars) in AZD7648-treated cells and a near-complete absence of indels (red bars).
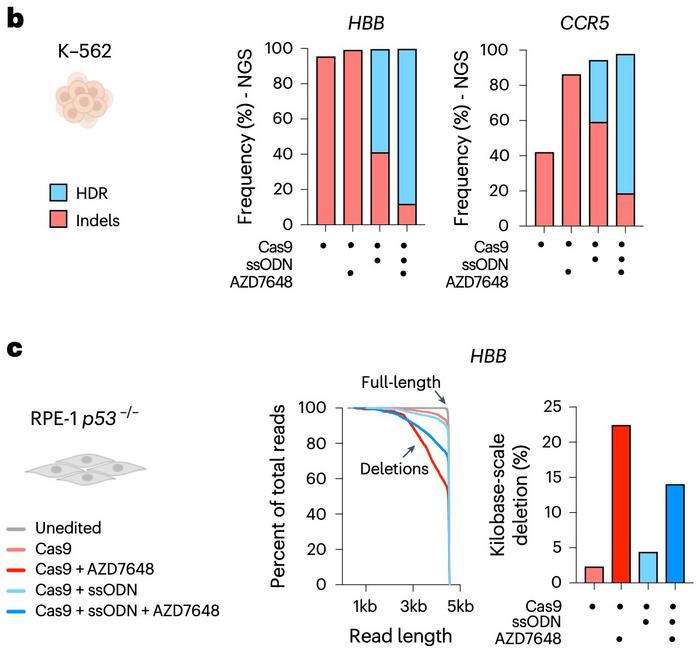
However, when the gene-editing outcomes were analysed by long-read sequencing (see Figure 1c), the results shifted dramatically. The inclusion of AZD7648 revealed that a substantial fraction of alleles carried kilobase-scale deletions (dark red and dark blue) – edits so large they had effectively vanished from the short-read data.
»When we used assays that looked exactly at the site of the cut, then we could see lots of HDR alleles and nothing else,« Corn sums up. »But when we used loci that reported on how many alleles there were total outside the cut, those alleles were also going away. Basically, the HDR alleles were sticking around because they weren't getting destroyed.«
In other words, anything that wasn’t HDR was being lost – and standard analyses had simply failed to catch it.
This suspicion was further reinforced by a fluorescent reporter system designed to distinguish between different repair outcomes. Depending on whether the break was repaired by indels or HDR, the system would emit different colours. Surprisingly, some of those signals disappeared entirely in cells treated with AZD7648. This finding suggested that the entire reporter sequence had been deleted – not just modified.
»The sequencing would imply that all the lights had changed colour. But when we actually looked, we saw that the candles were getting blown out entirely,« explains Corn.
Chromosome arms are lost down to the telomere
Having established that kilobase-scale deletions were common, the team next turned their attention to even larger structural changes. If short reads could miss deletions of a few thousand base pairs, what else might be escaping detection? To answer that, they moved beyond sequencing alone and began probing the genome at a much broader scale.
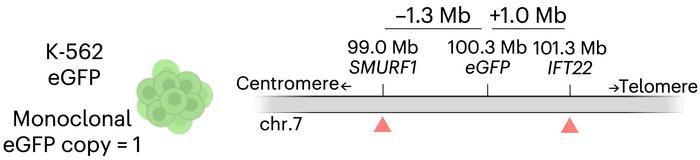
Corn, Cullot and colleagues engineered a monoclonal K-562 cell line carrying a single, known copy of the eGFP gene integrated on chromosome 7 (see Figure 2a). Then, they used CRISPR-Cas9 to introduce a cut at a site 1.3 megabases upstream of the eGFP insertion, with or without AZD7648. If the genome remained intact, cells would still express the fluorescent reporter. But if something catastrophic happened between the cut and eGFP, the cells would lose their signal. And that’s exactly what they observed.
Flow cytometry revealed that up to a third of the AZD7648-treated cells had gone dark. Further analysis using digital droplet PCR confirmed what the researchers suspected: the reporter gene wasn’t just silenced – it was gone. In some cells, the deletions appeared to extend all the way down the chromosome arm. As illustrated in Figure 2b-c, this combination of genomic setup, fluorescence loss, and copy number quantification clearly pointed to megabase-scale deletions.
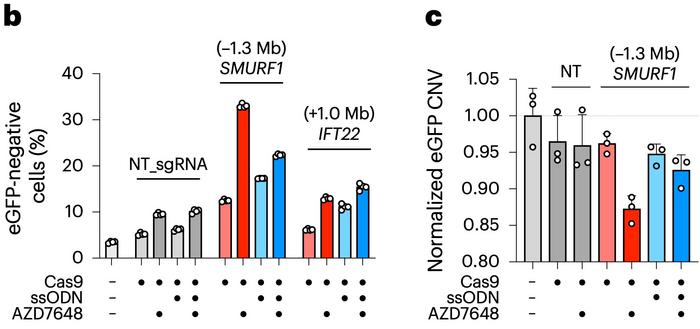
»Instead of just losing megabases away, you would lose like the entire rest of the chromosome arm all the way down to the telomere,« Corn explains.
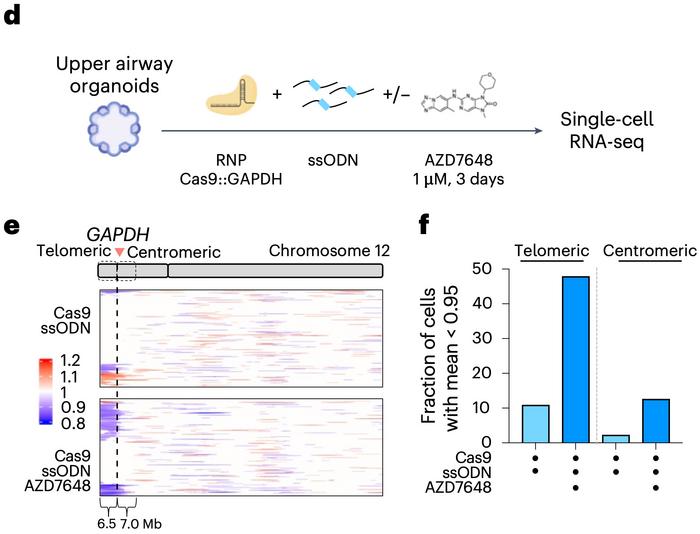
But the researchers didn’t stop at cell lines. To assess whether these large deletions also occurred in more clinically relevant models, they turned to human upper airway organoids and CD34+ haematopoietic stem and progenitor cells (HSPCs). These primary cell systems are not only more representative of therapeutic contexts but also less tolerant of genomic instability – making them ideal for uncovering potential risks.
Editing was performed in the 3′ untranslated region of the GAPDH gene – a site chosen to minimise any direct impact on cell health. Using single-cell RNA sequencing, the team then looked for patterns of gene expression loss across the genome. The results were striking. In the organoids, nearly half of the cells exhibited a loss of expression across a 6.5-megabase region extending from the cut site to the telomere. A similar, though somewhat less pronounced, effect was observed in HSPCs.
AZD7648 might have significant clinical implications
These patterns, presented in Figure 2d–f, suggest that megabase-scale deletions and chromosome arm loss are not just artefacts of immortalised cell lines – they occur in primary cells, too. Crucially, they happen at frequencies that could have significant implications for therapeutic genome editing.
While the team did explore ways to mitigate the damage caused by AZD7648, success proved partial at best. Many of the kilobase-scale deletions showed features typical of microhomology-mediated end joining (MMEJ), so the researchers tested whether blocking this pathway with a polymerase theta inhibitor, PolQi2, could help.
Indeed, combining PolQi2 with AZD7648 reduced the frequency of kilobase-scale deletions while preserving elevated HDR levels. But it did nothing to stop the megabase-scale damage. As Corn puts it, »We were excited – we thought, ah, great, now we can be back in business. But the thing that’s weird is that it didn’t help the megabase-scale deletions at all.«
That finding may have raised more questions than it answered. If kilobase- and megabase-scale deletions respond differently to interventions, they likely arise through distinct cellular pathways. For Corn, that’s both a challenge and an opportunity.
»I think a lot of the field would assume these deletions happen the same way. But this result implies that they don’t, and that’s pretty interesting,« he speculates.
Perhaps most importantly, the study reminds us of how research should work – rigorous, open-minded, and cautious in its optimism. AZD7648 does increase HDR, but at a cost that can’t be ignored. Several groups evaluating it for clinical use have already backed away after seeing the data.
»This is one case where research worked the way it’s supposed to,« says Corn. »New data came in, and now they’re probably not going to use it anymore – unless there's a way around it.«
Link to the original article in Nature Biotechnology:
To get more CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.
ArticleInterviewNewsHomology-directed Repair (HDR)Next gen sequencingOrganoidsSafety