FDA Clears ARCUS Editor for Duchenne Hot-spot
CMN Clinical
Feb. 18, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Universal T cell products for the treatment of cancers and infectious diseases seemed an unattainable goal. Now, researchers at Kyoto University's Center for iPS Cell Research and Application (CiRA) have made the breakthrough many clinicians and patients have been waiting for. Dr Wang, the lead author of the study, joined Professor Shin Kaneko's lab at CiRA in 2014 as a PhD student to use induced pluripotent stem cells (iPSCs) to generate universal T cell products for the treatment of cancers and infectious disease. His work, published in Nature Biomedical Engineering demonstrates how allogeneic T cells can be generated from gene-edited iPSCs to evade the host immune response while retaining their tumour-killing capacity.
T cell therapies involve the knock-in of either chimeric antigen receptors (CARs) or antigen-specific T cell receptors (TCRs). These modifications can be made to autologous cells from the same individual or to allogeneic T cells taken from healthy individuals – the problem, Dr Wang says, is that despite their potential, both strategies have significant trade-offs.
For autologous T cell therapy, there is no risk of immune rejection, however, generating the product is labour-intensive, and the financial and temporal constraints of personalized medicine are a major drawback.
»There is great potential for the use of autologous T cells to treat patients, but it's very high cost, with lengthy production times, and these factors really limit the large-scale clinical application of the cells,« Wang remarks.
While allogeneic T cell therapies are less expensive, the risk of graft versus host disease (GvHD) or graft rejection is high. Genetic modification and clonal expansion of T cells is difficult because edits often induce dysfunction or exhaustion. It is also problematic to exclude cells with off-target edits or mutations. For Dr Wang and his colleagues, the obvious solution to these problems was to use T cells derived from iPSCs, which are much more amenable to genetic modification.
»We wanted to utilize iPSC technology because they can be easily clonally expanded, and it's straightforward for us to perform gene modification in these cells, even multiple gene modifications,« Wang says
There are several factors at play in the host immune response to transplanted allogeneic T cells.
To create hypoimmunogenic T cells, Wang first needed to elucidate how host immune cells recognize and reject their transplanted counterparts. One of the key contributors is the human leukocyte antigen (HLA) system, including HLA Class 1 (HLA-I) and HLA Class 2 (HLA-II), which are expressed on the surface of T cells. The host T cells will recognize non-self HLAs, causing immune rejection.
Looking in detail at the allogeneic rejection, the host CD8 T cells recognize the mismatch of HLA-I on the transplanted T cells, and the host CD4 T cells recognize non-self HLA-II.
Another component is the activation of natural killer (NK) cells, which occurs if the grafted T cells exhibit low expression of NK cell inhibitory ligands like Bw4, C1 or C2, or overexpression of NK cell-activating ligands.
»We needed to eliminate all of these activating factors, so we did step-by-step gene modification,« Wang explains.
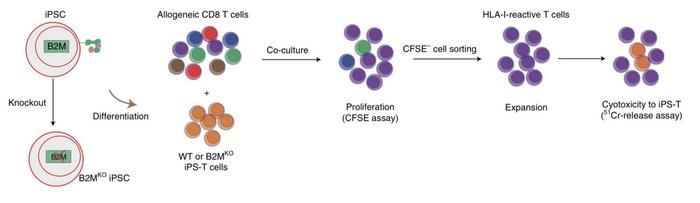
First, they co-cultured wild-type iPSC-derived T cells with allogeneic CD8 T cells, which induced the proliferation of CD8β reactive cells. Then they knocked out the β2-microglobulin (B2M) gene in the iPSC-derived T cells, which is essential for HLA-I formation.
»If we co-culture these B2M knockout cells with allogeneic CD8 T cells, there is no T cell proliferation,« says Wang.
After successfully evading the response of host CD8 T cells, Wang and the team turned to the next obstacle - the CD4 T cells. To prevent the expression of HLA-II on the iPSC-derived T cells and evade the CD4 T cell response, they targeted the Class II transactivator (CIITA) gene.
»We see that B2M knockout cells evade CD8 T cells. However, they still elicit proliferation and activation of CD4 cells, so we performed a knockout of CIITA to eliminate HLA-II expression in the cells. If we co-culture the B2M and CIITA knockout cells with allogeneic CD4 T cells, they no longer induce proliferation and activation of CD4 T cells,« says Wang.
At this stage, the double-knockout iPSC-derived T cells still activated host NK cells. However, controlling this NK cell response proved to be the biggest hurdle for the team.
»To control NK cell activation, we focused on the single-chain HLA-E (scHLA-E) molecule, which is known to be an NK cell inhibitory ligand. Then, using a lentiviral vector, we transduced scHLA-E into the double knockout iPSC-derived T cells,« Bo Wang says.
This approach reduced the response from the NK cells but did not completely eliminate it, so the team needed to go back to the drawing board.
To figure out what was causing the NK cells to respond – even in the presence of an inhibitory ligand – Bo Wang and his colleagues screened the double-knockout iPSC-derived T cells for NK cell activating ligands to determine which of them might be the culprit.
»We found that when the T cells are active, they upregulate the Poliovirus receptor (PVR, CD155). This is an activating ligand for the NK cell receptor DNAM-1. So we hypothesized that if we knocked out PVR in our double-knockout iPSC-derived T cells, maybe we could reduce the NK cell response further. We were correct - after we deleted this gene, the NK cell activation was significantly decreased,« Wang describes.
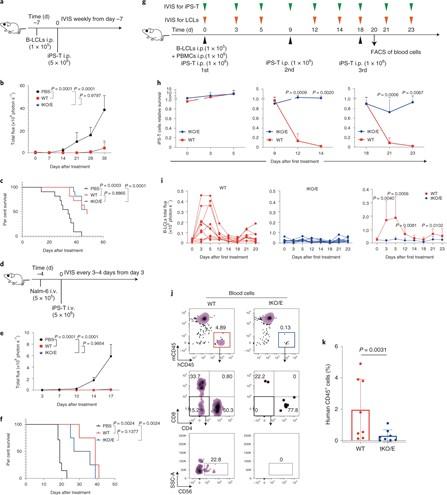
Triple-knockout iPS CAR T (tKO/E iPS-T) survival and tumour progression control experiments in a human immunocompetent mouse model. (A–C) tKO/E iPS-T cells mediated the control of B-LCL in vivo. (A) Experimental design. B-LCL-pre-inoculated mice were treated with tKO/E iPS-T cells on the indicated schedule. (B) The progression of B-LCL cells was measured by bioluminescence. (C) The Kaplan–Meier curve shows the percentage survival of the experimental groups. (D-F) tKO/E iPS-T cells mediated the control of Nalm-6 in vivo. (D) Experimental design. (E) The progression of Nalm-6 cells was measured by bioluminescence. (F) The Kaplan–Meier curve shows the percentage survival of the experimental groups. (G) Experimental design for H, I. (H) Relative survival ratio of the injected iPS-T cells at the indicated day in the presence or absence of PBMCs. (I) B-LCLs bioluminescent quantification of mice treated with WT or tKO/E cells. (J, K) Flow cytometry (J) and quantification of human immune cells (K) in iPS-T cell-injected mice. For a detailed description, see Wang et al., Nature Biomedical Engineering (2020) [Figure 6].
Wang had now successfully generated triple-knockout iPSC-derived T cells. Still, two critical questions were left to answer, and the answers would determine whether Wang's engineered cells would have any translational capacity.
The first was whether the cells were truly hypoimmunogenic and could survive in the presence of the complete host immune system. The second was whether the cells had retained their ability to control the progression of tumours.
To answer the first question, the team co-cultured allogeneic peripheral blood mononuclear cells (PBMCs) alongside either wild-type iPSC-derived T cells or triple-knockout iPSC-derived T cells.
»At day zero, the PBMC and wild type cells are detected, but at nine days, we can hardly detect wild-type iPSC-derived T cells because they don't survive in the presence of PBMCs - there is a much higher proliferation of CD8, CD4 and NK cells, which reject the wild-type iPSC-derived T cells,« says Wang.
In comparison, even after day nine of co-culture, they can still detect the triple-knockout cells – they didn't induce much proliferation of CD8, CD4 and NK cells. This indicates that the edited cells dramatically decrease immunogenicity and can survive even in longer cultures with the complete immune system.
To determine the survival and therapeutic potential of the edited iPSC-derived T cells compared to wild-type iPSC-derived T cells in vivo, the team turned to an immuno-deficient mouse model.
They co-transplanted human PBMCs and Epstein–Barr virus-transformed B-lymphoblastoid (B-LCLs) cells expressing CD20+ into the mouse, mimicking tumour progression. Then the mice were treated with either wild-type iPSC-derived T cells or the triple knockout cells, both of which expressed CD20 chimeric antigen receptor (CAR).
»We checked the survival of the cells in the mouse model over three treatments. On day two after the second injection, the wild-type cells are barely detected. But the triple-knockout cells can still be detected at a very high level. Moreover, even two days after the third treatment, the triple-knockout cells still survive,« Bo Wang says.
“Our triple knockout CAR T cells control tumour progression very well, just as well as the wild-type CAR T cells”Bo Wang
It was the final in vivo experiment that was the moment of truth. Once the team knew the triple knockout iPSC-derived T cells could survive in an allogeneic setting, they wanted to check if they could control tumour progression in the mouse model and the unedited CAR T cells.
»Our triple knockout CAR T cells control tumour progression very well, just as well as the wild-type CAR T cells,« Wang says.
Wang's study provides a substantial contribution to the field of T cell immunotherapy. Not only are the engineered iPSC-derived T cells hypoimmunogenic, meaning that they are a genuine 'off-the-shelf' product that could be used to treat any patient, but they can also be used to treat multiple disease states, including various cancers and infectious diseases.
The next step, of course, is to test the technology in a pre-clinical model to gain approval for clinical trials, a process Wang asserts is already underway in the Kaneko lab.
»We will try the triple-knockout iPSC-derived T cells to treat GPC3-expressing cancer. This means transducing the cells with a GPC3-recognizing CAR, which we already have in our lab. I can't say a detailed timeline, but maybe within five years, we will hopefully reach the clinical trial stage,« Bo Wang says.
Wang is optimistic that trials will be successful and that his breakthrough will be widely used.
» It's our goal to create a universal, off-the-shelf T cell therapy that can be used on both infectious disease and cancer, on potentially any patient. This paper shows the great potential for that,« Wang says.
Link to original article in Nature Biomedical Engineering:Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells.
Rebecca Roberts is a molecular biologist and science writer/communicator based in Queensland, Australia.