FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Trials that involve the use of CRISPR, base editing, and other gene-editing techniques to improve CAR T-cell therapies, including one currently recruiting in the UK, are focusing on paediatric patients with relapsed or refractory B-cell acute lymphoblastic leukemia, with the potential to expand to other indications in the future. Professor Waseem Qasim tells us here about his group’s work within this area and where the field stands as a whole.
- What makes T cells an attractive target for gene-edited therapies?
There’s a longstanding experience with T cells from the bone marrow transplant setting. And you can collect them reasonably easily from peripheral blood, and you can take them to a clean room and do things them — engineer them, manipulate them, characterise them, you can freeze them, and thaw them and give them back, so that makes them attractive.
When we come to gene editing, we now need to do other things including electroporation steps to get, for example, CRISPR reagents into the cells, or the RNPs [ribonucleoproteins] and guides. And they tolerate that reasonably well — I mean, they don’t like it, they get upset, and some will die through that process, but you can recover them, you can expand them, and then you can freeze them back down again.
- Where does your group stand on the various trials using edited T cells? You started with TALENs [transcription-activator-like effector nucleases] a few years ago, correct?
It’s still very early days. Yes, in London we have done the first TALEN-edited CAR T-cells, and that we reported back in 2015. That had gone remarkably quickly from laboratory development into the clinic; they had only been described a few years before that, TALENs, and the ability to then manipulate them and use them for a useful purpose was fairly rapid.
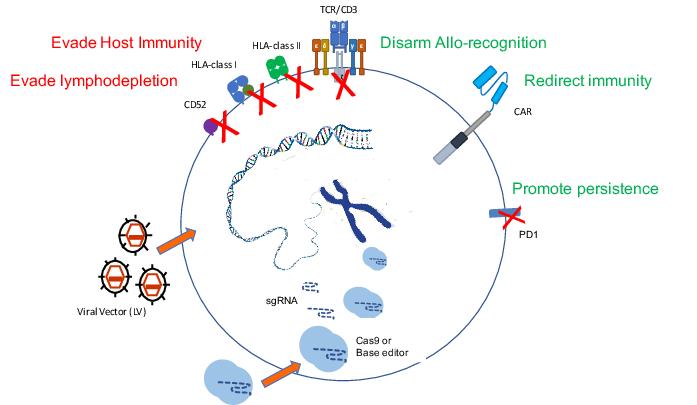
We did learn quite a lot, and are still learning, from those original trials about what it takes to engineer T cells that are otherwise allogeneic, meaning not matched to a particular recipient, and how difficult it is to actually overcome the barriers we all have to HLA immunity. We did two things to those cells: we removed the T cell receptor, and instead of removing Class 1 or Class 2 MHC, we removed something called CD52, which was the antigen that would otherwise render them susceptible to alemtuzumab, one of the conditioning antibodies.
[With these phase I trials], if things are looking like they’re going to work, we’re expecting companies to pick them up or take them on for development, and that’s exactly what happened with the TALEN version.
[Ed. note: alemtuzumab, a monoclonal antibody used to treat chronic lymphocytic leukemia and some other malignancies, targets B and T lymphocytes expressing CD52, so T cells engineered to lack CD52 would have a survival advantage in the presence of the drug, thus enriching the TCR-free cells' activity.]
- And from there you fairly quickly moved on to CRISPR?
As soon as the TALEN study had opened, we realised that the CRISPR platform was going to be more flexible from an academic point of view, and set off making a CRISPR version of T cells that are edited for those same sites, TCR and CD52. And that’s taken us about three years, and it’s now ready, and we’ve manufactured enough cells to service the Phase I study. The study is open in the UK but it has been held up with the COVID issues obviously, but it is open now and we’ll start recruiting children in the same situation [with relapsed/refractory B-cell acute lymphoblastic leukaemia]. The study is aiming to treat 10 children in a Phase I setting. At the moment we’re only open for children, but we’ll see if that changes.
- What are the challenges or obstacles inherent to using this approach?
Generally, with the use of CAR T-cells, from the beginning, there have been worries about the cytokine storm, the neurotoxicity, so the infrastructure around managing all those side effects has to be in place in the hospital setting. In the UK, the NHS has been quite good to make sure it’s being offered in centers that can offer all of that.
As the experience builds up, it’s becoming more familiar, so the side effects and the profiles are being understood. From the point of view of these versions, the allogeneic versions that we’re trying to make universal, I think we have a good understanding now that the barrier actually is quite high. Overcoming GVHD [graft-versus-host disease] I think has turned out to be more straightforward — you remove the T cell receptor and GVHD risk goes down. Overcoming the host’s ability to reject is more of a challenge.
There’s no free lunch, there’s always something in the formula that needs to be accounted for.
- So even though the gene-editing approach allows CAR T-cell therapy to move away from a “bespoke” treatment tailored to each patient separately and toward an off-the-shelf option, there are still aspects that are very patient-specific?
In reality I don’t think these issues are insurmountable, they will be overcome, because the same issues are there if you have an autologous product — you still need to be given some degree of lymphodepletion, you still have to tolerate the cytokine issues and the neurotoxicity and everything.
And the ability to do it off-the-shelf, I think, could change the dynamic of how we do it and when we do it, whether it’s brought earlier into the hierarchy of treatments, as the experience builds up. We are just at the outset here, it’s very early days, and the numbers need to build up for the experience to be understood across the different patient groups.
- So far you’ve focused on paediatric patients with B-cell malignancies. What other settings do you anticipate might make it to clinical trials in the near future using gene-edited T cells?
We’ve got patients with unmet need around other forms of blood cancers — myeloid leukaemias in particular. These are very difficult to treat if they don’t respond to the first rounds of treatment, or if they go to transplant and they relapse it’s then very difficult to treat those patients and they almost inevitably end up in palliation, so that’s one group we’re building towards the next round of trials.
And for those, in fact, we’ll move from CRISPR to base editing, because we’re doing multiplex editing and we like using base editing there because the risk of translocations between different edited sites drops off.
And then there are the T cell malignancies themselves where we’re trying to use CAR T-cells to treat T cell cancers. Up until now that has been very difficult because of the issue of fratricide, it’s very hard to make products because they kind of fight each other and consume each other in the culture period. So we can address that with the editing steps.
Those two targets in particular are for us the next goals — AML, and T-ALL in children. We’re in a stage where we’re trying to translate those into a manufacturing phase now, so we’re probably 12 or 18 months away from trying to get trials under way in those particular settings.
Waseem Qasim’s lab at Great Ormond Street Institute of Child Health at University College London investigates novel treatments and therapeutic approaches to treat various types of cancer. In particular, they focus on the use of T cells and how to marshal their effects to better fight off the malignancies. Along with collaborators at other academic institutions and with pharmaceutical and biotechnology companies, they help lead early-phase clinical trials using approaches such as TALENs, CRISPR, and base editing, in particular in pediatric patients who often lack effective treatment options. Their work encompasses the full range of therapeutic development, from lab bench to bedside, and their hope is that the treatments they develop in these early trials, if promising, will be moved forward by other collaborators to become useful options for many patients.
- Could you explain a bit more about the base editing approach?
Again that’s come on remarkably fast. We were tinkering with CRISPR, and David Liu published his first base edit [in 2016]. Within 18 months we have it in a configuration that we can take to a scale-up and try and manufacture cells.
It’s using a deactivated Cas element to take an enzyme to a particular location in the genome. A good application seems to be to use it to knock out genes — to introduce stop codons or to disrupt splice sites to knock out expression of particular genes. And we can do that as efficiently as CRISPR editing.
We don’t get the double-stranded breaks, or the risks of translocations between multiple edit sites, so that’s probably a plus. The risk of transformation in T cells is considered low, but we like the idea of not stressing that system — fewer translocations must be a good thing, it can’t be a bad thing.
The field is not standing still, it’s evolving rapidly. If we have this discussion again in 18 months’ time, I don't know what we’ll be using then.
- I imagine the speed of the field must be somewhat daunting.
It always was a bit daunting because you’re taking things into clinic for the first time, and you’ve got to think about the safety issues of that, and the risks and benefits.
But the speed at which the reagents have evolved, especially in the last four or five years, has been incredible. And it’s hard keeping up with all the laboratory developments, taking them into a translational step, and then a clinical step. We’re having to cover the whole spread of activity from bench to bedside, and the speed at which those things are moving is very fast.
- What’s your outlook for the field in a general sense, looking ahead the next few years?
Most of our applications have been through ex vivo engineering of cells. There’s obviously a massive interest in trying to get things to work in vivo directly. There’s a whole AAV [adeno-associated virus] field to try and deliver genes to do that. Whether AAV can meet the expectations or the requirements for many of these things I don’t know, we’ll have to wait and see; it’s not trivial doing some of those things.
In terms of the actual technologies, well, prime editing is on our radar, I’m sure there will be versions of that technology that work more efficiently or in a better way than the ones described so far.
If you’re coming through grad school or just graduating now looking for PhD topics, this has gotta be red hot for people looking for things to do. Job-wise down the line there should be lots of stuff going on. I think it’s a great time.
Dave Levitan is a science journalist based in the U.K.
ArticleInterviewDiseaseAcute Lymphoblastic Leukemia, ALLAcute Myeloid Leukemia, AMLCAR-TBase editorsCRISPR-CasTALENsGreat Ormond Street Hospital for Children NHS Trust





