
Intellia Files BLA After Lonvo-z Meets Phase 3...
CMN Clinical
Apr. 29, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Since 2012, CRISPR has been recognised as one of the most promising and flexible tools for manipulating genetic information. As a result, much research power has focused on improving CRISPR efficacy and safety, and in less than ten years we have seen a rising number of human CRISPR gene-editing clinical trials.The impressive speed with which this novel technology has been translated into therapies is also the result of effective targeting strategies.
Here we describe four current targeting strategies with CRISPR clinical trials as a case study and show why the targeting strategies are essential and how they are instrumental in developing present and future therapies to address genetic conditions at their roots.
First off, there are a couple of important considerations regarding the intended genetic modification to apply. First, the gene-editing intervention should not create unbalances in other functions of the cells, neither directly by perturbing genes involved in multiple facets of cell physiology nor indirectly by introducing unintended genetic modification known as off-targets. In other words, genetic editing should be as 'clean' and precise as possible.
Second, this makes it important to be aware that CRISPR gene-editing relies on the DNA repair mechanisms activated when the CRISPR-Cas9 endonuclease introduces a double-stranded break (DSB) in the DNA.
Very briefly, mammalian cells use two primary DNA repair mechanisms: the error-prone non-homologous-end joining (NHEJ) and the error-free homology-directed-repair (HDR) pathways. As it happens, mammalian cells prefer NHEJ to HDR, meaning that the majority of clinical applications involving CRISPR so far have focused on NHEJ.
Figure 1 gives an overview of the four different strategies, where the first three panels (a-c) are NHEJ, and the last (d) is HDR.
With these considerations covered, let us take a look at each of the four different targeting strategies.
CRISPR is a binary system, which relies on the Cas9 endonuclease to introduce a DNA double-stranded break (DSB) and a single guide RNA (sgRNA), which directs the Cas9 towards the desired target. The target is defined by a 20 nucleotide (nt) sequence present in the sgRNA called the spacer. When the sgRNA finds a sequence complementary to the spacer it will trigger the endonuclease activity of the Cas9. As a result, it is possible to reprogramme CRISPR to target any genetic location within the genome by simply changing the spacer sequence of the sgRNA.
One of the first CRISPR-based methods focuses on the inactivation of the faulty gene associated with the disease to be treated.
Inactivation can be accomplished by directing CRISPR to the gene's coding DNA regions called exons. Once a DSB occurs, most cell types will repair it via the error-prone NHEJ, typically introducing insertions or deletions (indels). These may alter the proper reading frame and create a premature stop codon resulting in a shorter transcript. It may be degraded (non-sense mediated decay), or the corresponding protein will be more unstable and immediately committed to the proteasome for degradation (Fig.1 a).
One example where this strategy has been applied is against the Human Immunodeficiency Virus (HIV). HIV primarily infects CD4+ T cells by entering via the C-C chemokine receptor type 5 (CCR5).
The presence of a 32-bp deletion (known as Delta32) within Exon1 in approximately 10% of the European population results in CCR5 inactivation and confers natural resistance to HIV infection.
Because of this, a knock-out of the CCR5 gene in HIV patients' derived CD4+ T cells is an appealing gene-editing strategy. The strategy was first explored more than ten years ago using Zinc Finger Nucleases and more recently also using CRISPR.
Let's take another example of how CRISPR mediated gene knock-out can be exploited, this time in cancer therapy. Notably, the laboratory of CAR T pioneer Dr Carl June has developed CRISPR to simultaneously knock out three genes to render the CAR T cells more efficient at battling cancer. One of the targeted genes is PD-1, which cancer cells often exploit to evade immune system surveillance. In the first published study, the engineered CAR T cells were administered to three patients – one man and two women – who experienced no side effects after receiving the CRISPR gene-edited cells, alas also without any benefit. You may find more details in a dedicated article here and a similar clinical trial led by Professor Lu You and Professor Toni Mok that was published shortly after, which also demonstrated the safety of CRISPR-edited T cells in 22 patients with aggressive lung cancer.
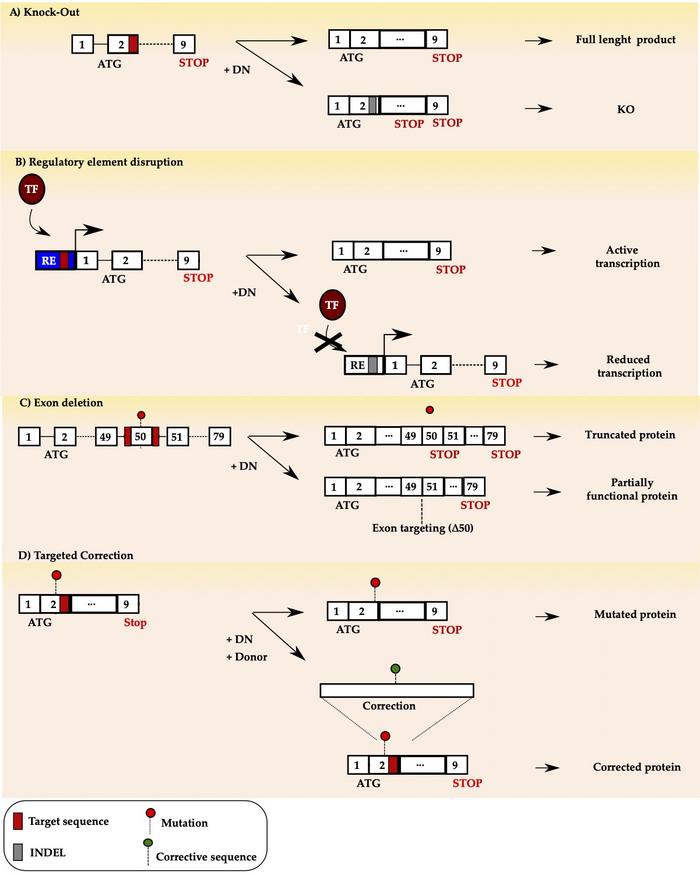
Schematic illustration of the four gene editing strategies using designer nucleases (DN). a) targeted knock-out b) regulatory element (RE) disruption to prevent rranscription factor (TF) binding c) exon skipping d) targeted correction.
Sometimes targeted knock-out may not be an effective strategy and may even be detrimental for the patient. This is the case in sickle cell disease (SCD) and β-thalassemia (BT).
Both conditions share a defect in adult haemoglobin synthesis caused by mutations in the HBB locus. Previous research indicated that foetal haemoglobin could help individuals with SCD or BT by compensating for defective adult haemoglobin. In adults, foetal haemoglobin expression is suppressed, favouring the adult counterpart by a transcription factor encoded by the BCL11A gene. However, a targeted knock-out approach is not recommended when the whole cell physiology would be jeopardised in the attempt of aiding a specific condition.
The BCL11A protein is a transcription factor expressed in different haematopoietic lineages. Thus, its function is not only restricted to control switching from foetal to adult haemoglobin. Furthermore, since the lack of BCL11A expression results in defects in HSCs, its knock-out would be a counterproductive strategy.
An attractive strategy, therefore, could be to disrupt the BLC11A erythroid-specific enhancer region. Mutations in this region correlate with elevated foetal haemoglobin levels without abrogating BCL11A expression in the other haematopoietic lineages.
This is the strategy adopted in recent successful clinical trials to treat SCD and TDT, which led to the reactivation of foetal haemoglobin to therapeutic levels with no reported side effects (Fig.1 b).
As a result, patients who received the treatment are no longer expected to require transfusions to keep their symptoms under control, and SCD patients are no longer subjected to painful vaso-occlusive episodes due to the presence of sickle-haemoglobin. You can read more about these exciting developments here.
Some conditions may require a more complex approach, like Duchenne muscular dystrophy (DMD), a genetic disorder characterised by progressive muscle degeneration and weakness caused by a mutation in the dmd gene.
DMD is the largest gene in the human genome, with a 14 kb mRNA transcript that exceeds the packaging capacity of standard viral vectors, making gene replacement approaches challenging to apply. Instead, it is possible to programme CRISPR to disable the mutated exon's splice acceptor (SA) or splice donor (SD) sequence. This approach aims at restoring the DMD transcript's reading frame by excluding – or skipping – the mutated exons. Skipping may result in a shorter – but still functional – protein, which could help to alleviate DMD and progress to a milder form of the disease, similar to Becker muscular dystrophy (BMD). Exon skipping is not a new strategy as it has been attempted using antisense oligonucleotides (AOS). However, contrary to AOS, CRISPR offers the possibility of one-shot-only treatment as it acts at the DNA level rather than at the transcript level (Fig.1 c).
Even though no clinical trials have been conducted for exon skipping so far, the increasing number of promising in vivo studies suggests that it won't be long before CRISPR is used to demonstrate its effectiveness in a first clinical trial.
Finally, treating the condition may require precise gene editing rather than its targeted disruption. In this case, CRISPR gene editing will rely on the endogenous HDR pathway rather than the NHEJ pathway. Particularly, a donor DNA containing the desired modification is provided together with CRISPR. The aim is to trick the cell into exploiting the HDR path to use the supplied DNA donor as the blueprint to repair the DSB introduced by the CRISPR-Cas endonuclease.
This strategy is particularly amenable when correcting conditions derived by point mutations. However, HDR-based approaches are hampered by the fact that cells use NHEJ preferentially over HDR and that non-cycling cells do not use HDR to repair DSB.
However, research groups such as that of Dr. Matthew Porteus at Stanford University have put a lot of effort into improving HDR-based approaches by optimising AAV vectors to deliver the donor DNA. The latest results in this direction come from a recent publication where they could achieve a high level of corrected long-term HSCs using CRISPR combined with an AAV6 donor, paving the road for future clinical trials.
On the other hand, the rise of DSB-free editing platforms, which do not require HDR to introduce precise modifications, may bypass these limitations. In particular, recently base editing was successfully used to correct the mutation responsible for Progeria in a mouse model. Thus, even though NHEJ-based therapies are prevalent now, things may change in the near future thanks to CRISPR-based technologies such as base editing and prime editing.
The presence in the genome of sequences sharing a partial similarity to the desired CRISPR target may result in undesired genetic alterations, the so-called off-targets. Off-targets may generate aberrant chromosomic rearrangements as well as altering oncogene expression. Because of this, off-target profiling is a pivotal aspect of clinically oriented CRISPR applications. To date, several detection tools have been developed, each of them having pros and cons. However, they all have in common a PCR amplification step of the targeted locus using the genomic DNA of the edited cell pool as a template. The PCR amplicons are then used for further downstream analysis. You can find a more detailed description of this topic in this recent interview with Dr Eric Paul Bennet.
CRISPR is applied in clinical trials to treat genetic diseases at their source by using different targeting strategies: targeted knock-out, regulatory element disruption, exon skipping and precise genetic correction. In each of these four solutions, CRISPR is adapted to remove, correct or bypass the defective gene while retaining the remaining genetic information.
It is still early days for the CRISPR gene-editing therapies and the number of CRISPR gene-editing clinical trials is growing fast.
A cure for the first previously incurable genetic diseases such as sickle cell disease seems within reach soon. New platforms like prime editing and base editing are emerging rapidly as well. All told, the CRISPR progress so far has been incredibly uplifting to address the range of genetic disorders long thought to be incurable.
Antonio Carusillo, PhD, currently employed at Vector BioPharma.





