
Intellia Files BLA After Lonvo-z Meets Phase 3...
CMN Clinical
Apr. 29, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...
CRISPR-Cas is often seen as as the holy grail in medicine, with hopes that it will offer cures for for rare genetic diseases with poor or no treatment options. Here, we take a look at the current status of CRISPR gene editing for the treatment of Duchenne muscular dystrophy (DMD).
DMD is a rare muscle disease but it is one the most frequently encountered genetic disorders, occurring in approximately 1 in 3,500 to 5,000 live male births worldwide.
DMD is an X-linked disease that arises from mutations in the DMD gene that encodes dystrophin (see Fact Box). Dystrophin is a cytoplasmic protein that plays a mechanical function in muscle. By tethering the actin cytoskeleton to the plasma membrane of the muscle cell, dystrophin protects and maintains muscle integrity during normal activity and exercise.
Mutations in the DMD gene lead to a shortage of dystrophin, resulting in the hallmark signs of the disease, such as progressive muscle weakness and muscle death, inflammation and loss of mobility before adulthood. Respiratory and cardiac dysfunction occur in advanced stages of the disease, and affected individuals usually die prematurely between the 2nd and 4th decade of life.
A number of treatments that slow disease progression through partial dystrophin restoration or modulation of inflammatory processes have been approved for clinical use, but as of yet, no cure exists.
Since DMD is caused by mutations that lead to dysfunctional dystrophin variants, pharmacological approaches to treatment provide no hope of a cure since they cannot address the genetic defect. However, these are still important in disease management.
A number of corticosteroids such as prednisolone are used to help maintain muscle strength by reducing inflammation and increasing muscle bulk. Steroids can slow disease progression, and they are currently the only medicines available to all DMD patients.
Other treatments include physical therapy, dietary supplementants and medications to manage other problems that may arise as disease progresses, e.g., cardiac, respiratory or digestive issues.
The DMD gene is the largest human gene, consisting of 79 constitutive exons and a significant amount of intronic sequence that collectively spans over 2.2 MB of genomic DNA.
At least 17 wild-type DMD transcript variants are known, and one or more of these are expressed and translated in all types of muscle as well as various other cell types.
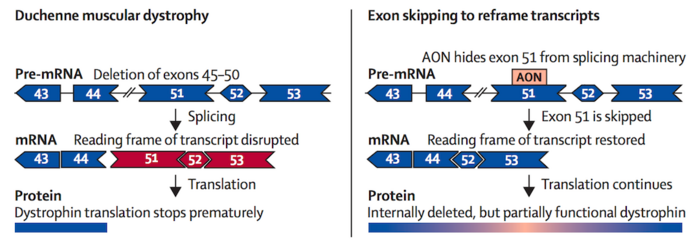
Thousands of disease-causing mutations have been reported for DMD, ranging from single point mutations and deletions to large deletions or duplications of one or more exons. While mutations can be found anywhere along the gene, deletion of one or more exons is the most common defect. Exon deletion oftens disrupts the reading frame, preventing the remaining exons from splicing together properly, resulting in dysfunctional dystrophin variants.
Disease severity is very variable, and depends on the location and size of mutations, whether or not the mutation(s) are in-frame or out-of-frame (out-of-frame mutations lead to truncated dysrophin variants that lack the C-terminal and are generally associated with severe disease), and whether the mutations affect muscle tissue globally or only the cardiac muscle.
Disease severity can also vary for the same mutations, sometimes even within the same family. The sheer number of mutations and the lack of correlation between mutation and disease phenotype makes DMD a very challenge disease to target.
The overall number of treatments available to DMD patients is very small, but novel genetic approaches that address the underlying genetic cause of disease provide fresh hopes.
In recent years, four gene-targeting therapies have received FDA approval for DMD. All of these are antisense olignonucleotide (ASO) treatments that work by binding to the pre-mRNA dystrophin transcript. These function as molecular patches that force the translation machinery to skip certain exons in order to restore the reading frame to yield albeit truncated dystrophin variants that retain some function. Such variants may maintain muscle integrity and produce a less severe disease phenotype.
The approved therapies, eteplirsen (exon 51, Sarepta Therapeutics), golodirsen (exon 53, Sarepta Therapeutics), viltolarsen (exon 53, NS Pharma) and casimersen (exon 45, Sarepta Therapeutics) each induce skipping of a single exon and may be used to treat patients with mutations in exons 45, 51 or 53, which account for about 30 % of all DMD cases.
As of yet, the European Medicines Agency has not approved any of the ASOs, but it did approve ataluren (PTC Therapeutics), a premature stop codon readthrough therapy to treat paediatric DMD caused by nonsense mutations in 2014.
While further advances in therapeutic exon-skipping strategies can theoretically address more than 80 % of DMD mutations, there have been some concerns about their efficacy so far, they do not address cardiac dysfunction in DMD, and this class of treatments is not curative.
Emerging strategies to develop multi-exon skipping therapies through conjugation with muscle-homing peptides to ASOs may increase the proportion of eligible DMD patients as well as increase efficacy by improving the cellular uptake and longevity of the oligonucleotides in the target tissue. Efforts in these areas have shown promise so far in dogs, mice and cell lines derived from DMD patients. However, a number of regulatory challenges and safety concerns will have to be addressed before such strategies are approved.
Late last year, Sarepta Therapeutics announced positive clinical data from its Phase 2 MOMENTUM trial for SRP-5051, a peptide-conjugated antisense-oligonucleotide designed to target exon 51 with increased tissue penetration and efficacy.
As a single-gene (or monogenic) disease, DMD is a prime candidate for gene-editing therapy and it was among the first monogenic diseases to be investigated for CRISPR-based gene correction.
An in vivo CRISPR therapy would address DMD mutations at the genomic level, thus offering a permanent correction and bypassing the need for regular dosing.
The programmable nature of the Cas endonucleases also makes it feasible to target sequences throughout the DMD gene by changing the guide RNA and repair template as necessary to theoretically address any intended mutation, thus broadening the scope of targetable mutations.
For instance, CRISPR can used to mimic the mechanism of exon skipping in a more efficient “one and done” manner by deleting certain exons permanently to restore the DMD reading frame. It can also be used to delete duplicated exons in the DMD gene to restore the gene sequence completely and permanently.
Although technically more difficult, owing to DNA donor template size limitations and the low efficiency of homologous recombination in muscle cells (because they are post-mitotic), it is also possible to knock missing exons in to the DMD gene to restore expression of a full-length intact dystrophin protein.
Beyond the gene-editing capabilities of CRISPR mentioned above, the related base-editing technology offers therapeutic potential for the estimated 25-35 % of DMD cases that involve point mutations in the DMD gene. Moreover, catalyically dead forms of Cas9 (dCas9) have recently been fused to transcriptional activators or repressors to create fusion proteins that can be programmed with a guide RNA to switch on or off gene expression at desired genomic locations.
The potential of CRISPR to treat and even cure DMD is very exciting, and although no CRISPR-based therapies have entered clinical development yet, very encouraging findings have emerged from research in this area. Some of the significant findings will be mentioned here.
In 2019, researchers from University of Texas Southwestern Medical Center (US) along with Exonics Therapeutics, which was later acquired by Vertex Pharamceuticals, demonstrated that systemic administration of AAV-delivered CRISPR-Cas9 and gRNAs could correct mutations in exon 44 in DMD patient-derived cells and a mouse model harbouring the same mutations. The same researchers previously demonstrated correction of exon 50 mutations in mice and also in dogs.
Research led by Charles Gersbach at Duke University in 2016 deployed systemic or local AAV delivery of CRISPR-Cas9 machinery to delete mutated exon 23 from the DMD gene of mdx mice, resulting in the expression of a semi-functional dystrophin variant and improvements in muscle function. The same group previously used single or multiplexed gRNAs to target the common mutational hotspot at exons 45–55, restoring human dystrophin expression in patient-derived cells in vitro and in vivo following transplantion into mice. mdx mice are a popular model for DMD, and they contain a single point mutation in their DMD gene, which results in a premature stop codon and non-functional dystrophin.
Last year, research from Germany revealed that CRISPR-Cas9-mediated deletion of exon 51 in pigs lacking exon 52 resulted in widespread dystrophin expression in muscle, including diaphragm and heart, extending survival and reducing the risk of irregular heartbeat.
So far, attempts to correct DMD mutations in patient-derived cells as well as mouse, dog and pig models of DMD have shed encouraging results.
CRISPR offers distinct advantages over exon-skipping therapies and other gene-targeting approaches not discussed here, such as RNA editing and full-length gene replacement therapies, of which the latter have been superseded by surrogate gene therapy attempts to deliver smaller micro-dystrophin alternatives with partial functionality.
The possibility to permanently correct mutations with a single CRISPR treatment would in principle prevent further disease progression without the need for repeat dosing. If efficient enough, gene editing may also restore close to or even normal levels of dystrophin expression under endogenous regulatory control, thus ensuring normal temporospatial expression patterns.
As with all other gene-editing therapies, concerns exist about off-target edits that may lead to undesired dystrophin variants or disrupt other regions of the genome. Advances in the CRISPR field more broadly, including the development of high-fidelity Cas endonucleases and improved gRNA design, as well as the emergence of new delivery strategies, continue to push boundaries with respect to safety and gene-editing efficiency.
Beyond the "standard" CRISPR challanges, there are also a few unique caveats to gene correction for DMD. Since DMD is a progressive disease, no amount of gene correction will be able to restore the damage already done by lost muscle function. Therefore, the best therapeutic outcomes will be acheived with treatment early in the course of disease. Whether CRISPR can indeed cure DMD even when adminstered early remains to be seen, but neonatal testing will at least identify cases early in life.
Efficient systemic delivery of gene-editing cargo to all muscle tissue including the heart is no mean feat. AAV delivery has thus far been a promising approach in research settings and on a small scale, but translating this to clinical scale for the large number of DMD patients in need will be very challenging. Furthermore, the potential for long-term dystrophin expression following gene editing remains to be shown, and this is related to both the muscle turnover rate and the rate of gene-editing efficiency. If muscle turnover and regeneration occur via non-edited satellite cells, the effect of gene correction will likely diminish over time, and the immunogenicity of AAV vectors precludes a repeat treatment with the same AAV serotype. Moreover, it is also not yet known what percentage of muscle cells must be corrected to achieve a therapeutic benefit in humans.
All of these open questions and considerations must be investigated before CRISPR reaches the clinic for DMD, but with the rate the CRISPR field is moving, there is reason to be optimistic. At present, Vertex Pharmaceuticals, Editas Medicine, CRISPR Therapeutics and Sarepta Therapeutics are working on CRISPR programmes for DMD, all of which are at the preclinical stage. We look forward to bringing you updates on these programmes as they advance towards clinical stage.
Literature Sources Used
ArticleAdenovirus (AV)Duchenne Muscular Dystrophy, DMDRare DiseaseGene therapyCRISPR-CasCas9dCas9Sarepta Therapeutics





