Inhaled CRISPR-Cas9 nanoparticles disrupt Sting1...
CMN Briefs
Mar. 9, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...
CMN WEBINAR - Development of CRISPR/Cas9-based Therapies against Alzheimer's Disease
Speaker: Martin Ingelsson, Prof., University Health Network, Toronto, Canada
On-demand webinar available here

Although disrupting pathogenic mutations in amyloid precursor protein (APP) has been proposed as a therapeutic strategy for patients with Alzheimer’s disease (AD, see Fact Box), little progress has been made in the treatment of familial AD by targeting mutations associated with early-onset disease.
In a recent study, a team of researchers led by Professor Martin Ingelsson at the Department of Public Health and Caring Sciences at the Rudbeck Laboratory, Uppsala University (Sweden), reported using CRISPR-Cas9 to selectively disrupt the mutant allele PSEN1M146L in human fibroblasts, which led to a reduction in the extracellular Aβ42/40 ratio and restoration of PS1 levels and conformation.
»CRISPR-Cas9 treatment could partially restore the AD-related Αβ42/40 ratio seen in the mutant cells from AD patients, while off-target effects were minimal,« said Evangelos Konstantinidis, the first author of the study. »Most PSEN1 mutations are associated with increased generation of the more aggregation-prone Aβ42 peptide, and our approach mitigates that phenotype,« he added.
Konstantinidis also noted that, as most mutations leading to early-onset AD display an autosomal dominant inheritance pattern, the team believe that targeting PSEN1 mutations at an early stage could prevent the progression of Aβ pathology and disease onset.
The team's findings were published in Molecular Therapy Nucleic Acids.
Alzheimer’s disease (AD) is a neurodegenerative disease and the most common cause of dementia worldwide. Although sporadic late-onset AD is relatively common, familial early-onset AD is rare, accounting for less than 5% of all AD cases. Mutations in PSEN1, the gene encoding presenilin 1, result in an increased Αβ42/40 ratio and are the most common cause of familial AD.
“CRISPR-Cas9 treatment could partially restore the AD-related Αβ42/40 ratio seen in the mutant cells from AD patients, while off-target effects were minimal. Most PSEN1 mutations are associated with increased generation of the more aggregation-prone Aβ42 peptide, and our approach mitigates that phenotype.”Evangelos Konstantinidis
Presenilin 1 (PS1) is one of the four protein subunits of the enzymatic complex γ-secretase, a protease that mediates the cleavage of various transmembrane proteins. By promoting the generation of amyloid-β (Aβ) peptides from APP, γ-secretase can contribute to the accumulation of Aβ plaques in the brain, which is a key manifestation of AD. Therefore, PS1 has emerged as an attractive therapeutic target for AD treatment.
Several mutations in PSEN1 (gene encoding PS1) have been shown to predispose individuals to AD by altering the enzymatic function of γ-secretase and enhancing the formation of amyloid plaques.
Replacement of methionine with leucine at position 146 (M146L) of the PS1 protein is associated with the development of familial AD. The PSEN1M146L mutation leads to the increased generation of β-amyloid-42 (Aβ42), a particularly aggregation-prone form of Aβ peptide. Consequently, carriers of the PSEN1M146L allele develop familial early-onset AD with an average age at onset of 43 years and autosomal dominant inheritance.
Patients with AD carrying the PSEN1M146L allele show an increased Aβ42/Aβ40 ratio in the brain and other tissues; however, the underlying mechanisms remain unknown.
»We hypothesised that targeting the PSEN1M146L allele using CRISPR-Cas9 might help reverse AD-associated phenotypes by reducing the accumulation of extracellular Αβ deposits,« said Konstantinidis.
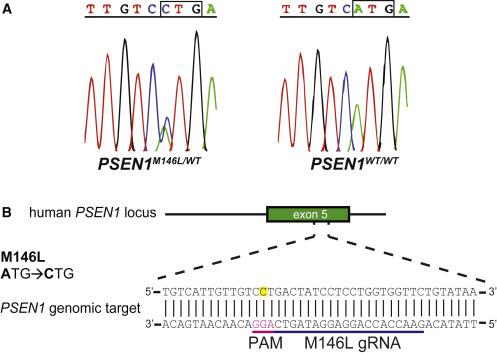
The team developed a CRISPR-Cas9-based method to selectively disrupt the pathogenic PSEN1 M146L mutation in human fibroblasts from heterozygous individuals (PSEN1M146L/WT) (Figure 1). To achieve this, they designed a gRNA targeting the A-to-C mutation in the PSEN1M146L allele.
Konstantinidis explained that this mutation is ideal for CRISPR-mediated targeting because it creates a novel PAM site in the reverse DNA strand that is absent in wild-type PSEN1: »Previous studies have shown a low preference of SpCas9 for 5′-NTG-3′ PAM sites like the one in the PSEN1WT allele, giving us confidence that the M146L gRNA would only disrupt the PSEN1M146L allele.«
After co-transfecting human fibroblasts with CRISPR plasmids encoding SpCas9 with either M146L-specific gRNA or scramble gRNA (control), the team performed next-generation sequencing (NGS) to confirm the disruption of the PSEN1M146L allele. They then evaluated the Aβ42/40 ratio and PS1 conformation to assess the potential of this CRISPR-Cas9-based strategy to treat patients with early-onset AD carrying the PSEN1M146L allele.
After selecting cells stably expressing the CRISPR plasmids, which made cells resistant to the antibiotic puromycin, the team performed NGS analysis of genomic DNA isolated from human fibroblasts from six carriers of the PSEN1 M146L mutation and four age-matched healthy individuals (two from the same family and two unrelated volunteers) to assess the efficiency of CRISPR-Cas9-mediated gene editing.
They found that PSEN1M146L/WT fibroblasts expressing SpCas9 and M146L-targeting gRNA exhibited indels in PSEN1 at the M146L mutation site. No indels in PSEN1 were detected in PSEN1WT/WT fibroblasts expressing SpCas9 and M146L-targeting gRNA or in PSEN1M146L/WT fibroblasts transfected with scramble gRNA.
Analysis of the percentage of allelic disruption demonstrated that indels were introduced in approximately 68% of the PSEN1M146L alleles in PSEN1M146L/WT fibroblasts expressing SpCas9 and M146L-targeting gRNA.
Commenting on the NGS results, Konstantinidis said: »These findings suggest that our CRISPR-Cas9 based approach can be used to selectively disrupt PSEN1M146L in cells carrying the PSEN1 M146L mutation.«
Most PSEN1 modifications introduced in PSEN1M146L/WT fibroblasts after treatment with CRISPR-Cas9 were frameshift mutations around the cleavage site (94.3%), while only 5.7% of modifications were in-frame mutations.
To determine the effects of CRISPR-mediated PSEN1M146L disruption on PS1 levels and conformation, Konstantinidis and colleagues performed western blotting and fluorescence resonance energy transfer (FRET)-based conformation analyses.
They found that PSEN1M146L disruption resulted in a ~40% reduction in the PS1 N-terminal fragment (p<0.01) and ~15% reduction in the PS1 C-terminal fragment (p<0.001) in PSEN1M146L/WT fibroblasts; however, PS1 levels remained unaltered in CRISPR-treated PSEN1WT/WT fibroblasts.
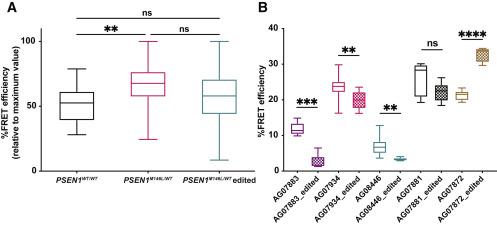
The team also found that the relative %FRET efficiency was significantly higher in untreated PSEN1M146L/WT fibroblasts than in untreated PSEN1WT/WT fibroblasts (~70% vs. ~50%, p<0.01), suggesting that PS1/γ-secretase had a tight rather than a relaxed conformation in cells harbouring the PSEN1 M146L mutation (Figure 2).
Although CRISPR-mediated PSEN1M146L disruption did not significantly affect the overall PS1 conformation in the pool of PSEN1M146L/WT fibroblasts, significant relaxation in PS1 conformation was observed in three out of five samples of PSEN1M146L/WT fibroblasts.
»Our FRET-based conformation analysis data suggest that CRISPR-mediated disruption of the PSEN1M146L allele may restore the conformation of PS1 by altering the distance between the C- and N-terminal domains of the protein or by reducing the levels of the mutant protein,« Konstantinidis explained.
Interestingly, one sample showed a significant increase in %FRET efficiency after PSEN1M146L disruption (~40% vs. ~20%, p<0.0001), suggesting that targeting the PSEN1 M146L mutation resulted in tighter PS1 conformation in this sample. “This finding really surprised us,” Konstantinidis said. »We hypothesise that this finding may be related to the fact that the ‘outlier’ sample was the only one taken from an older individual who already had clinical manifestations of AD.« He explained that the patient’s condition and age might have affected the amount of intracellular mutant PS1 present in that sample.
The team analysed the levels of Aβ42 and Aβ40 in the culture medium of human fibroblasts to evaluate the effects of PSEN1M146L disruption on the extracellular Aβ42/40 ratio. As expected, non-edited PSEN1M146L/WT fibroblasts had a significantly higher extracellular Aβ42/40 ratio than non-edited PSEN1WT/WT fibroblasts (~0.14 vs. ~0.09; p<0.05). Notably, CRISPR-mediated PSEN1M146L disruption in CRISPR-Cas9-treated PSEN1M146L/WT fibroblasts reduced the extracellular Aβ42/40 ratio from ~0.15 to ~0.12 (p<0.01).
»These results suggest that CRISPR-mediated PSEN1M146L disruption can only partially restore the extracellular Aβ42/40 ratio, as the Aβ42/40 ratio was higher in CRISPR-edited PSEN1M146L/WT fibroblasts than in wild-type cells,« Konstantinidis noted, who then explained that this finding might be attributed to differences in gene-editing events among the treated samples, which they analysed as a pool rather than as individual cell clones because of difficulties in expanding single-cell clones.
Although the findings suggest that targeting PSEN1M146L at an early stage could prevent AD onset, Konstantinidis noted that the study is still at a very early stage and a more thorough assessment of the effects of CRISPR-Cas9 editing should be conducted.
The team now plans to confirm its findings in single-cell clones of edited cells. »We believe that the observed phenotype would be completely normalised if we were able to generate fibroblast colonies from single cells. In that way, we could have selected the ‘most edited’ cells for further analysis. However, we also think that this approach made our analysis more ‘real-life-like’, given the heterogeneity that occurs when providing CRISPR components in vivo.«
Konstantinidis also said that, to achieve a higher editing percentage, they plan to stably express Cas9 and gRNA in fibroblasts by transducing them with adeno-associated viruses or lentiviruses. Future plans also include the use of iPSC-derived neurons from patients carrying the PSEN1 M146L mutation to analyse treatment effects on a broader panel of phenotypic alterations.
Dr Rudolph Tanzi, an expert in AD research, noted that there are practical and ethical challenges to overcome before we are able to use CRISPR to treat different forms of AD. He is the Joseph P. and Rose F. Kennedy Professor of Neurology at Harvard University and did not participate in the study.
»It is relatively straightforward to use genomic editing to correct a gene defect and its consequences in cells in a Petri dish. However, the challenges to clinically achieving this for a gain of function gene mutation such as this one, in all relevant neurons in the brain, would be both medically and ethically challenging,« he said.
»We realise that our approach is still at a very preliminary stage, but this type of research could bring us one step closer to developing efficient therapies for AD and other neurodegenerative diseases,« said Konstantinidis.
Link to the original article in Molecular Therapy Nucleic Acids:
Christos Evangelou, Ph.D., is a freelance medical writer and science communications consultant.
To get more of the CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.