FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Dr Celine Rocca, from the Cherqui Lab at the University of California San Diego’s School of Medicine, has been trying to develop a treatment for Friedreich’s ataxia for some time. Friedreich’s ataxia (FRDA) is a rare, multisystemic neurodegenerative disorder, occurring in approximately 1 in 40,000 people.
In 2020, Rocca and her colleagues published a breakthrough paper in Molecular Therapy: Methods & Clinical Development, describing the use of CRISPR to edit patient-derived haematopoietic stem and progenitor cells (HSPC) to treat FRDA. In a recent interview, Rocca explained the genetic basis for the disease, the need for precision models of FRDA, and the significance of her study to the FRDA community.
Friedreich’s ataxia is an autosomal recessive disease caused by mutations in the FXN gene, which codes for the mitochondrial protein frataxin. Frataxin is involved in several key processes in iron metabolism, antioxidant protection, and energy production in the mitochondria. FXN features a trinucleotide repeat in its first intron; in a healthy individual, the gene typically contains between five and 30 GAA repeats. In patients with FRDA, however, FXN contains a hyper expansion of 44 to 1700 GAA repeats.
This hyper expansion reduces the production of the frataxin protein, resulting in the accumulation of iron in the mitochondria and subsequent oxidative stress. As a result, the condition causes degeneration of sensory nerve fibres in the dorsal root ganglia (DRGs), affecting the brain, spinal cord, heart, skeletal muscle, and peripheral nerves, leading to progressive loss of muscle coordination, strength and sensation. Speech, hearing and vision are all affected by FRDA. Many sufferers also experience cardiac dysfunction and, to a lesser extent, scoliosis and diabetes mellitus. Interestingly, the number of repeats is directly correlated with the age of onset and the severity of the condition.
Friedreich’s ataxia (FRDA) is a rare, multisystemic neurodegenerative disorder, occurring in approximately 1 in 40,000 people. Currently, there is no cure for FRDA, with treatment options limited to addressing associated illnesses in order to extend the lifespan of affected individuals. Within 15 years of diagnosis, the majority of FRDA patients lose the ability to walk and are wheelchair-bound. The condition is often fatal, with 59% of deaths in FRDA patients caused by cardiac dysfunction.
For pre-clinical research to progress, precision models of disease are critical. Unfortunately, until the advent of gene editing technologies – particularly CRISPR – a lack of translational models has been a major stumbling block for researchers hoping to develop novel treatments for diseases like FRDA. Mouse models of FRDA currently show limited translational capacity, as they lack the full complexity of neurodegeneration seen in humans with the disease. As Rocca explains, there were several options available, but all had significant limitations.
»For a previous study, we used the YG8R mouse model – this one gave us a very nice phenotype of the disease: It replicated what we see in human patients. But it has a duplicated expansion, which is not the same genotype as human patients. The same lab that developed the YG8R model later produced an improved version – YG8sR, which has only one expansion and is more genetically accurate, but we lose the disease phenotype. For HSPC transplantation for Friedreich’s ataxia, we need a mouse model that recapitulates the progression of the disease. We tried on a cardiac-specific knock-out model of Friedreich’s ataxia, but the phenotype was too strong – the mice die at only a few months old, so the stem cells don’t have the time to engraft and provide healthy mitochondria to the diseased cells. So it would help if you had a very progressive model to get the therapy to work,« she describes.
A recent study, published by Dr Vania Broccoli’s lab in the Neuroscience Division of the San Raffaele Scientific Institute, used patient-derived induced pluripotent stem cells (iPSCs) to generate DRG organoids as a precision model of FRDA. The researchers then used CRISPR to delete the first intron of FXN, where the hyper expansion is located, restoring the expression of the gene. This study is significant for several reasons: it provides a platform for modelling the disease, enables potential treatments to be tested, and provides supporting evidence for Rocca’s assertion that CRISPR can be used to treat FRDA.
For many diseases caused by the missing or reduced production of specific proteins, lentiviral mediated HSPC transplantation can be used to deliver healthy proteins to patients. One of the complicating factors in FRDA treatment, however, is that too much frataxin can have a toxic effect on the cells, so it’s critical that the production of the protein is carefully regulated.
In 2017, Rocca and her colleagues at UCSD tried transplanting wild-type mouse HSPCs into the YG8R mouse model of FRDA. The transplanted cells differentiated into microglia and macrophages, grafting in the affected tissues of the brain, spinal cord, DRGs, heart, and muscle, and restored healthy mitochondrial function. This was mediated by transferring the functional frataxin from HSPC derived cells to diseased cells through tunnelling nanotubes. The treatment arrested the development of muscle weakness and degeneration of sensory neurons in DRGs, mitigating the effects of the disease.
While the study successfully demonstrated the potential of allogeneic HSPC transplant in treating the disease, the team still needed a safer treatment for human patients.
»Allogeneic transplantation in human patients remains very risky because you have to find the perfect match donor, and there is a risk of rejection, so we thought about developing another strategy that would be safer for the patient. Usually, we use lentiviruses to correct HSPC ex vivo and provide the functional protein. But with Friedreich’s ataxia, it’s tricky because if you overexpress the protein, it can be toxic,« Rocca cautions.
Using CRISPR, researchers like Rocca and her colleagues can now edit the disease-causing mutation in the FXN gene and treat the condition via cell or gene therapy.
»We thought the proper way to restore physiological expression of the frataxin protein was to delete the GAA expansion in the first intron of the gene that’s responsible for the decreased expression of frataxin in the cells,« Rocca explains.
Fortunately, heterozygous carriers of the hyper expansion are asymptomatic, meaning that editing just one allele of the gene is sufficient to increase frataxin production and restore normal function in Friedreich’s ataxia patients. As Rocca adds, the hyperextension is also in a convenient location for gene editing.
»Friedreich’s ataxia is a perfect disease for a gene-editing approach because the expansion is within an intron, so you can cut almost wherever you want without introducing mutations,« she says.
The limitations of FRDA mouse models forced Rocca to use HSPCs taken directly from FRDA patients for the present study. Long-term, the team wanted to take HSPCs from the FRDA patients, correct the FXN mutation, and transplant the patients’ healthy autologous cells back to them. This strategy would, in theory, eliminate the risks associated with using donor cells and effectively treat the disease, as they had previously demonstrated in mice. But, first, they needed to prove that the cells could be edited to restore a healthy phenotype.
To edit patient-derived HSPCs using CRISPR, Rocca and her colleagues collaborated with Associate Professor Prashant Mali, a genome and stem cell engineering expert at UCSD’s Bioengineering department.
»We used a ribonucleoprotein (RNP) complex of the crRNA and the Cas9 protein. In addition, several guide RNAs for slightly upstream and downstream of the expansion were tested to see which gave us the best gene-editing in terms of the percentage of corrected cells,« Rocca elucidates.
Once the hyper expansion had been excised, the production of frataxin increased as expected. The only hiccup, Rocca notes, was an initial p53-mediated delay in cellular proliferation.
»There was a delay of a good 24-48 hours in the cell proliferation. This was described by another lab recently, so we knew it was expected – it’s an effect of creating the double-stranded breaks in the DNA. Some labs are now developing compounds to overcome that - a method of inhibiting p53 while you perform the gene editing. So that might be a way to overcome that issue in the future,« she says.
Luckily, after this initial delay, the cells began to divide normally, with no other long-term adverse effects.
Notably, the team noticed no off-target editing or cellular toxicity.
»There was no toxic effect, just a delay in proliferation. So we need to be very careful when we develop cell therapy for human patients that we are transplanting the cells at the correct time. We don’t want to transplant them in a state when they are not proliferating because this will decrease the engraftment,« Rocca observes.
»It was very specific as well – we observed no off-target editing, so we were fortunate.«
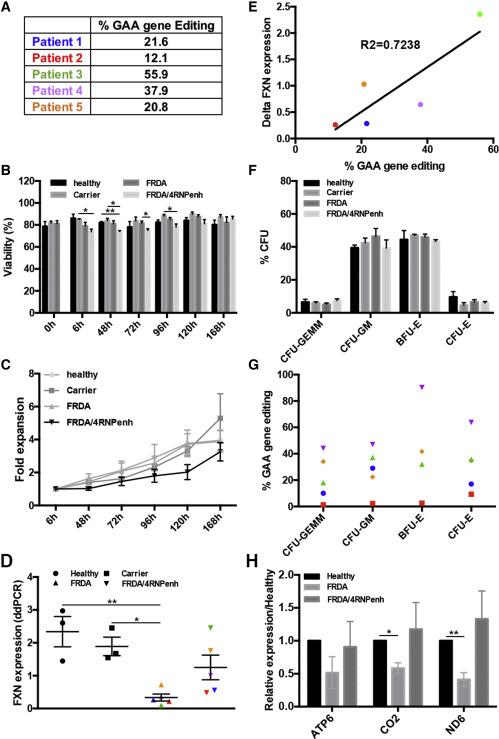
GAA gene editing in Friedreich’s ataxia (FRDA) patient cells and the impact on FXN expression. (A) GAA gene-editing % in five different FRDA patient donors. (B) Viability of healthy, carrier, FRDA, and FRDA/4RNPenh CD34+ cells over time. (C) Fold expansion of healthy, carrier, FRDA, and FRDA/4RNPenh cells over time. (D) Quantification of human frataxin mRNA in healthy, carrier, FRDA, and FRDA/4RNPenh CD34+ cells. (E) Correlation curve showing the significant relationship between the percentage of GAA gene editing and the increased expression of human frataxin mRNA in gene-modified CD34+ cells from FRDA patient donors. (F) Colony-forming unit assay showing the percentages of the different colony types formed. (G) GAA gene-editing percentage in each colony type for each FRDA donor. (H) Quantification of the human mitochondrial (mt) complex subunit mRNA mtDN6 (complex I), mtCO2 (complex II), and mtATP6 (complex V) in healthy, FRDA, and FRDA/4RNPenh CD34+ cells. Figure from: Rocca et al., Molecular Therapy: Methods & Clinical Development (2020).
Rocca’s study is a significant step forward for FRDA research, and she hopes CRISPR-edited HSPCs will become a more common vehicle for treating genetic disorders in the future.
»HSPC transplantations are a common therapy to treat blood disorders. But for multisystemic disorders like cystinosis or Friedreich’s ataxia, it’s not always obvious that this type of therapy can work. We want people to know that HSPCs can be an excellent vehicle to provide healthy proteins to the entire body. It’s a very cool mechanism if you think about it – you provide a patient with their HSPC cells, where you have corrected the mutation ex vivo. The cells will differentiate into macrophages or microglia that can engraft throughout the body to deliver the functional protein to diseased cells,« Rocca comments.
»It’s very important to know that this is a one-time therapy - we don’t need to transplant the cells more than once.«
Rocca has now left the laboratory in which she performed the study. However, she continues to work in the field of cell and gene therapy, now at Genethon, a non-profit organization dedicated to designing gene therapies for rare diseases. She is particularly interested in finding cures for muscular dystrophies using novel cell therapies and understanding how the HSPC derived cells and muscle fibre cells communicate with each other to find new ways of treating these diseases.
It’s difficult to say when clinical trials for FRDA will get underway, Rocca says. [CRISPR Therapeutics and Capsida Biotherapeutics just announced plans to develop a gene-editing therapy Friedreich’s Ataxia]. Still, the lack of cellular toxicity and absence of off-target edits in patient-derived HSPCs make her confident about the efficacy and safety of the treatment. The remaining members of the Cherqui lab are continuing with the work and are currently gathering pre-clinical data in a more accurate mouse model.
»Now there is a new model, with an increased GAA expansion, so the team has a closer genotype and phenotype to the one observed in Friedreich’s ataxia patients to work with before they get FDA approval,« Rocca explains.
The initial success of a similar clinical trial for cystinosis designed by the Cherqui lab makes Rocca optimistic about using HSPCs to treat FRDA and other mitochondrial disorders.
»What we hope is that [the success of the method in trials for cystinosis] will be the same for Friedreich’s ataxia, and that we can cure other mitochondrial disorders in the same way.«
Link to original article in Molecular Therapy: Methods & Clinical Development: CRISPR-Cas9 Gene Editing of Hematopoietic Stem Cells from Patients with Friedreich’s Ataxia.
Rebecca Roberts is a molecular biologist and science writer/communicator based in Queensland, Australia
ArticleInterviewNewsFriedreich’s ataxiaCapsida BiotherapeuticsCRISPR Therapeutics AG





