Inhaled CRISPR-Cas9 nanoparticles disrupt Sting1...
CMN Briefs
Mar. 9, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...
»In previous Cas9-efficiency work, we could calibrate datasets using overlapping gRNAs, but here we had too few overlaps,« explains Jan Gorodkin at the University of Copenhagen. He is co-senior author of the paper that was published today in Nature Communications. He adds: »By labelling each gRNA by its dataset of origin, we could train effectively on all datasets at once without forcing them into a single unified scale.«
CRISPR base editors represent a refinement of classical CRISPR-Cas9 technology, enabling researchers to change single DNA letters without cutting both strands of the double helix. These molecular tools combine a modified Cas9 enzyme with a deaminase that chemically converts one DNA base into another. Adenine base editors convert A·T base pairs to G·C, whilst cytosine base editors convert C·G to T·A. However, the technology presents a complication: not only the intended base but also nearby "bystander" bases within an approximately eight-nucleotide window may be edited, creating multiple possible outcomes for any given gRNA.
»We were surprised to find that the base editors evaluated in this study still have a large editing window, which can introduce unintended bystander edits,« notes Yonglun Luo, co-senior author of the paper, at Aarhus University, Denmark. »That underlines the importance of prediction tools that capture both efficiency and the full spectrum of editing outcomes.«
“By labelling each gRNA by its dataset of origin, we could train effectively on all datasets at once without forcing them into a single unified scale”Jan Gorodkin
Predicting which outcomes will occur and how efficiently they will occur has proven difficult. Existing prediction tools have been trained on individual datasets, each with inherent limitations, including diverse experimental platforms, different deaminase variants with distinct sequence preferences, and an abundance of low-efficiency gRNAs that skew model training. The fragmented nature of available data has meant that models perform well on data similar to their training set but struggle to generalise across different experimental conditions.
»Using datasets generated by different studies for training a better prediction model is challenging due to data incompatibility issues, caused by factors such as expression level of the base editor, different versions of base editors, and cell-type differences,« says Yonglun Luo. »Our dataset-aware training approach overcomes this challenge and enables more accurate prediction of base-editing gRNAs.«
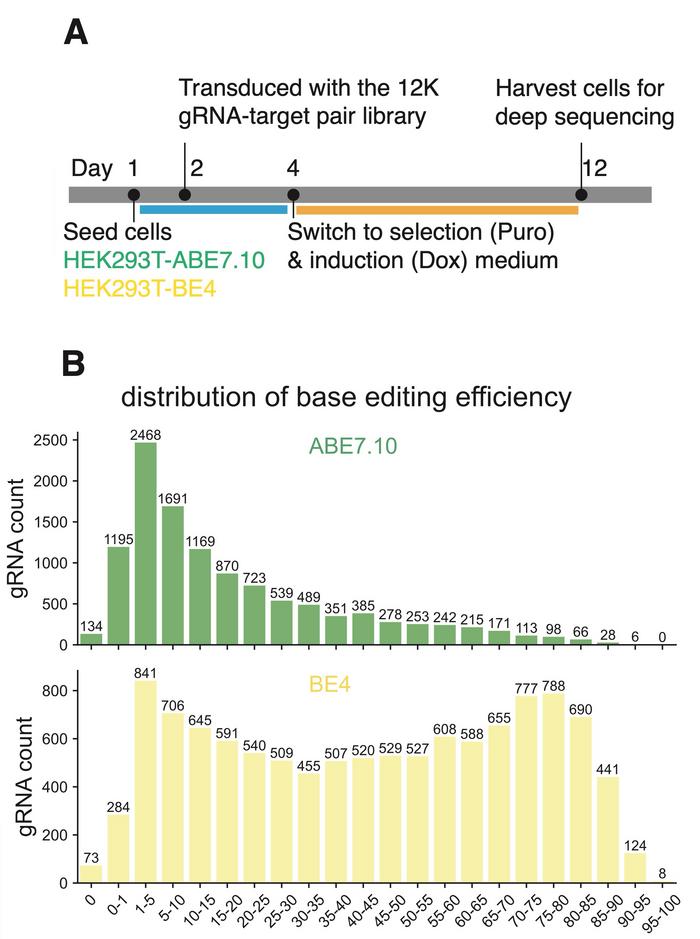
The research team addressed this by generating substantial new experimental data and developing a novel training strategy. Using their previously established SURRO-seq technology (see Figure 1), which creates libraries pairing gRNAs with their target sequences integrated into the genome, they measured base-editing efficiency for approximately 11,500 gRNAs each for ABE7.10 and BE4-Gam base editors in HEK293T cells. After quality filtering, they obtained robust measurements for over 11,000 gRNAs per editor.
Analysis revealed that ABE7.10 exhibited highly specific adenine-to-guanine transitions at 97%, whilst BE4 showed 92% cytosine-to-thymine specificity. Both editors displayed peak activity at positions four through eight in the protospacer sequence. Notably, the team found positive correlations between base-editing efficiency and standard Cas9-induced indel frequency at the same sites, suggesting that Cas9 activity predictions could inform base-editing models.
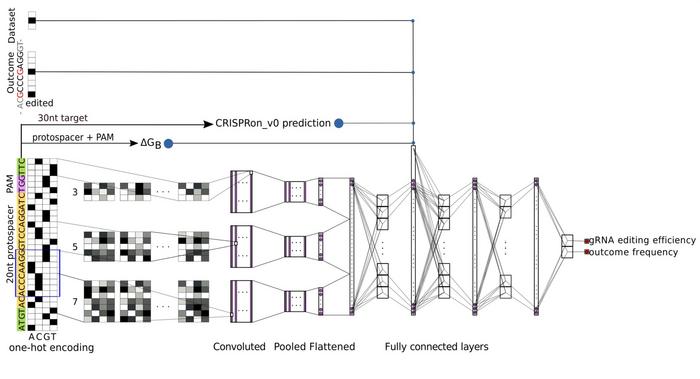
The key innovation lies in how they integrated their data with existing published datasets. Rather than simply pooling data, they developed an architecture that trains simultaneously on multiple datasets whilst explicitly labelling each data point's origin. For adenine base editors, they incorporated five datasets, including their SURRO-seq data and published data from Song, Arbab, and two Kissling datasets, examining both ABE7.10 and the engineered ABE8e variant. For cytosine base editors, they used three datasets from SURRO-seq, Song, and Arbab studies. The model architecture employs deep convolutional neural networks with multiple filter sizes to process the 30-nucleotide target sequence, alongside molecular features including gRNA-DNA binding energy and predicted Cas9 efficiency.
“We hope CRISPRon-ABE and CRISPRon-CBE will help other researchers select gRNAs giving the highest editing efficiency and the intended outcome”Yonglun Luo
The dataset labelling proves crucial. The team observed substantial heterogeneity across datasets, with the Kissling ABE8e data showing predominantly high editing efficiencies of 60% to 80%, whilst the Kissling ABE7.10 data for identical gRNA sequences showed lower efficiencies of 30% to 50%, reflecting the engineered enzyme's three- to eleven-fold increase in activity. By encoding the dataset origin as a feature vector, the model learns these systematic differences during training. Critically, users can make predictions which allow them to assign weights to different datasets, essentially tuning the model to match their experimental conditions.
The models, termed CRISPRon-ABE and CRISPRon-CBE, predict both overall gRNA editing efficiency and the frequency of specific editing outcomes simultaneously. To jointly evaluate performance on both metrics, the researchers introduce two-dimensional correlation coefficients that extend the standard Pearson and Spearman measures. Testing on independent datasets demonstrated consistent superiority over existing methods, including DeepABE/CBE, BE-HIVE, BE-DICT, BE_Endo, and BEDICT2.0. Feature analysis confirmed that predicted Cas9 efficiency plays an important role in base-editor predictions, and omitting dataset labels during training decreased performance by approximately 10%.
The researchers acknowledge limitations in their current models. They have incorporated only three base editor variants – ABE7.10, ABE8e, and BE4 – and all training data derive from HEK293T cells, except for datasets that include mouse embryonic stem cells and U2OS cells. Different deaminases exhibit distinct sequence motif preferences, meaning predictions for base editors not represented in the training data remain challenging. The dataset integration strategy could apply to other CRISPR technologies where data heterogeneity poses challenges. The models are available both as a web server and as standalone software under an academic use licence.
»We hope CRISPRon-ABE and CRISPRon-CBE will help other researchers select gRNAs giving the highest editing efficiency and the intended outcome,« says Yonglun Luo.
The study was led by Jan Gorodkin and Yonglun Luo at the University of Copenhagen and Aarhus University, respectively, in Denmark. The study was published today, 7 November 2025, in Nature Communications under the title "Deep learning models simultaneously trained on multiple datasets improve base-editing activity prediction".
The CRISPRon-ABE and CRISPRon-CBE tools can be accessed via https://rth.dk/resources/crispr/. Sequencing data is deposited in the China National GeneBank Database under accession CNP0001031 and NCBI Sequence Read Archive under accession PRJNA1321836.
To get more CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.
ArticleCMN HighlightsInterviewNewsOff-targetBase editorsSafety