CMN Weekly (6 March 2026) - Your Weekly CRISPR...
CMN Weekly
Mar. 6, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

After years of work, a massive team of scientists seems to have arrived at a one-time base editor gene therapy that can repair the point mutation causing Hutchinson-Gilford Progeria Syndrome, according to research published Wednesday in the journal Nature.
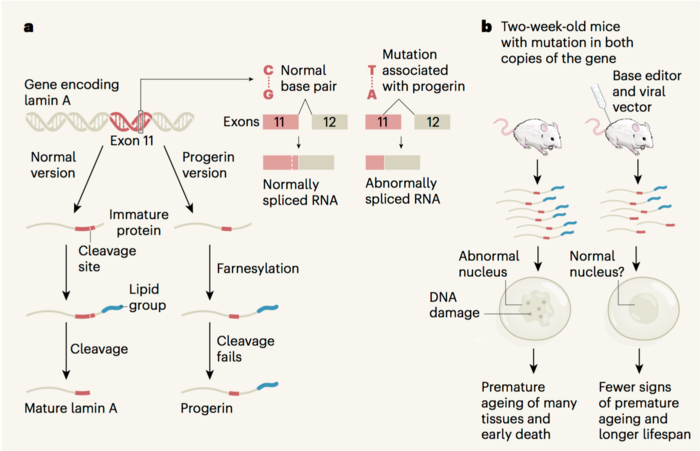
Hutchinson-Gilford Progeria Syndrome, often just called Progeria, is an extremely rare but fatal genetic disease. The disease stems from a randomly-occurring mutation, typically a dominant-negative CG-to-TA mutation in the Lamin A (LMNA) gene, which results in a truncated form of the protein progerin and ultimately causes cell death — to devastating ends.
The mutation leads to developmental issues and gives young children the appearance of having rapidly aged. The Progeria Research Foundation estimates that there are only about 350 to 400 Progeria patients alive — worldwide — at any given time, as the typical patient typically only lives about 14.5 years and Progeria isn't passed from parent to child or shared among siblings.
During those years, children tend to experience slowed growth, thinning skin and hair loss, skeletal abnormalities like fragile bones and joint problems, and most die of heart disease. In November, the U.S. Food and Drug Administration (FDA) approved the cancer drug lonafarnib as the first medication to directly treat Progeria, but there's still no actual cure for the disease.
Instead, clinicians have typically treated individual symptoms with physical therapy, steroids, dental work, hearing aids — the list goes on.
»Despite a very passionate community of patient families, doctors, and scientists, no treatment has yet proven to substantially extend the lifespan of children with the disease,« says study coauthor Dr. David Liu, a chemist at Harvard University, the Broad Institute of Harvard and MIT, and the Howard Hughes Medical Institute.
Finding a way to actually reverse that mutation, then, becomes a matter of urgency for the families of children with Progeria, who Liu says have formed a tight-knit and compassionate community.
Clinical research investigating the new genetic therapy is still a long way off, Liu says. But the study represents a major step forward on that front — and Liu was eager to give proper credit to the team effort.

Led by Luke Koblan, a chemical biology grad student in Liu's Harvard lab, the research was co-authored by prominent experts in the Progeria research community like Dr. Francis Collins of the National Institutes of Health — who bred a mouse model with the human form of the mutated LMNA gene — and Dr. Jonathan Brown of Vanderbilt University, in addition to other scientists who worked around the clock throughout the coronavirus pandemic.
In the actual study, the team treated that animal model — as well as cultured fibroblasts from children with Progeria — with an Adenine Base Editor (ABE). The ABE, delivered via a typical adeno-associated virus (AAV), targets the correct sequence in the LMNA gene and restores the mutant thymine base to a healthy cytosine.
»As you may know, base editors are engineered molecular machines we first developed in 2016 and 2017 to directly convert one DNA base pair at a specified position in the genome of a living cell into a different base pair, without cutting the DNA double helix (unlike earlier gene-editing agents such as ZFNs, TALENs, and CRISPR-Cas9, which primarily disrupt, rather than precisely edit, genes in most cell types),« Liu says.
About 90 per cent of the fibroblasts were corrected by the treatment, as were about 20 to 30 per cent of the mouse cells, depending on which organ you looked at. But even without correcting every cell, the treated mice showed a huge improvement over Progeria mice (see videos below).
Typically, Progeria patients will lose vascular smooth muscle cells in their heart, which is why heart disease is the leading cause of death among patients. But after six months, the aortas of ABE-treated mice were virtually indistinguishable from healthy mice when it came to cell count and phenotypic appearance.
The treated mice also looked generally healthier than saline-injected Progeria mice, who aged rapidly and died early — not unlike human patients with the disease. Compared to the average lifespan of 215 days for Progeria mice, the treated mice lived for approximately 510 days, which Liu explains is the mouse equivalent of reaching the start of old age. That means that the experimental gene therapy extended the mice's lifespan roughly 2.37 times over.
»Both of those were amazing to us and were the first sign that something very special was happening here, where a correction of a minority of the cells at the DNA level was leading to rescue that was much more profound at the organ level, and of course that would translate to a much longer lifespan,« Liu says.
»The 2.4-times longer lifespan is unprecedented in terms of any Progeria animal model. To have a 2.4-times longer lifespan is beyond what we could have hoped for.«
»Base editing has only been around for four years. If you had asked me even five years ago if we could use an engineered molecular machine to treat an animal model of Progeria and change a single mutation, I would have said 'Maybe 10 to 20 years from now',« says David Liu.
This approach is much more direct than lonafarnib — that's the treatment recently approved by the FDA — which doesn't reverse the mutation but instead tries to undo the damage to the progerin protein. Essentially, it chops off a farnesylated tail that's normally removed by a cleavage site that is itself removed from the Lamin A gene by the progeria mutation. So, the mutation still takes its toll, but lonafarnib seems to reduce the severity of the damage.
Using both lonafarnib and the new ABE therapy in concert, Liu speculates and hopes, may give patients an even better prognosis than either treatment does alone, but it's far too soon to say so with any confidence.
Clinical research on human Progeria patients is still a distant goal, but Liu's ultimate dream is to have the FDA approve the gene-editing therapy — or an improved version of it — as a one-time, postnatal injection.
In general, experts expect gene-editing therapies to work better the earlier in a patient's life they're administered. In the absence of prenatal testing for Progeria, Liu explains that his team's gene-editing therapy will need to be given after the patient is born and diagnosed. But because Progeria is a progressive disease, Liu expects — assuming the gene-editing therapy is proven to be safe and effective — that patients will see substantial benefits even if the treatment were administered late into the course of the disease.
»The Progeria patient community is so passionate, and they're quite well-connected. I'm hopeful that if this approach turns into a treatment, people will really know about it. And hopefully, if we can demonstrate that it's safe and efficacious in children, it will join lonafarnib as one of the only two treatments that might be available,« Liu says.
»The hope is that an affected Progeria child would get a one-time injection into their bloodstream — a base editor packaged into some kind of delivery vehicle that could then directly and permanently correct the root cause of the disease,« Liu adds.
»And, potentially, offer them a one-time treatment that could rescue at least some of the symptoms of the disease. That's the hope.«
But Liu was quick to reiterate that any assumptions about how well the treatment might work in human children was speculative — he very much wanted to avoid giving the Progeria community any false hope before he had the data to back it up.
To that end, he and the team are taking a two-pronged approach to further developing their treatment. On the one hand, they're pushing the current version, as described in their new paper, forward toward clinical trials. But they're also already in the early stages of a follow-up experiment that will treat mice at different ages and also test the various improvements to the base editor as well as parameters like the dosing levels that the team has made over the past several years.
All of these studies represent the very early stages of gene therapies for Progeria. But given the promising results for this new Nature paper, this ongoing endeavour will certainly be a project to keep an eye on.
»It was really a lot of work over a long period of time, and I feel so fortunate to have the opportunity to work with such a talented and dedicated group of collaborators,« says Liu.
Dan Robitzski is a science journalist and former neuroscientist based in Los Angeles.
Video of a Progeria mouse-model 7,5-month-old shows clear signs of premature ageing presented by co-author Urraca Tavarez, National Human Genome Research Institute, National Institutes of Health (NIH), Bethesda, USA, who worked through the Covid19 pandemic to care for and monitor the mice. Video courtesy of David Liu.

Video of Progeria mouse-model almost 10-months-old treated with CRISPR base editors to correct the disease-mutation. Compared to the untreated mice, these mice have shiny fur and are lively as normal mice at this age. Video presented by co-author Urraca Tavarez, National Human Genome Research Institute, National Institutes of Health (NIH), Bethesda, USA, who worked through the Covid19 pandemic to care for and monitor the mice. Video courtesy of David Liu.
ArticleInterviewMost readNewsAdeno-associated virus (AAV)Hutchinson-Gilford Progeria SyndromeGene therapyBase editors