FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Alessia Cavazza PhD joined Great Ormond Street Institute of Child Health (ICH) at University College London (UCL) in 2016. The institute is part of Great Ormond Street Hospital (GOSH), which has been a pioneer in gene therapy clinical trials for genetic immune disorders. Here, Cavazza became interested in studying the potential of emerging gene-editing technologies, e.g., CRISPR, to treat rare and incurable immune diseases such as severe combined immunodeficiency (SCID) and Wiskott-Aldrich Syndrome (WAS; Fact Box 1).
In 2017, Cavazza started her own group in the Section for Molecular and Cellular Immunology at ICH, and within just a few years the group successfully used CRISPR to correct the gene implicated in WAS. The results of the work were recently published in Nature Communications. We interviewed Alessia Cavazza to hear more.
»I started in the group of Professor Adrian Thrasher at UCL who led one of the first lentiviral-based gene therapy trials for WAS and who has studied WAS pathophysiology his entire career. We started discussing the trial results and about what could be improved. In general, lentiviral gene therapy works pretty well but there is great variability among patients and it seems to work poorly in the treatment of the platelet defects seen in WAS.«, explained Alessia Cavazza.
At that time, Alessia Cavazza’s main focus was developing a gene editing strategy for SCID, but a side project to develop a WAS knock-out cell line to study WASp function led her to shift her focus to WAS.
»I realised that the gene editing reagent I designed for WAS was working beautifully in primary haematopoietic stem cells, and this became my primary interest.«
Wiskott-Aldrich Syndrome (WAS) is an X-linked recessive primary immunodeficiency whose hallmarks include platelet defects, recurrent infections and complex immunodeficiency. WAS occurs as a result of mutations in the WAS gene which lead to defective WAS protein (WASp) expression and/or function. WASp is part of a large protein family whose members are critical for actin cytoskeleton function and maintenance. Unlike other family members, WASp expression is limited to the haematopoietic system and as such its deficiency only impacts blood cells. A healthy actin cytoskeleton is crucial for many cellular functions, such as cell morphology, migration, chemotaxis, receptor signalling, and cellular crosstalk. WAS deficiency impairs all of these functions in immune cells, leading to dysfunctional antibody-mediated responses and impaired immune synapse formation and T-cell activation after T cell receptor engagement. How exactly WAS mutations lead to platelet defects is less clear, but WAS platelets are smaller than healthy platelets and are functionally defective – properties which may accelerate their clearance by peripheral organs, resulting in the thrombocytopenia (low platelet numbers) seen in WAS patients.
Recognising the limitations of lentiviral gene therapy for WAS, Alessia Cavazza’s group turned to CRISPR to correct WAS in primary haematopoietic stem cells (HSPCs) isolated from WAS patients. The group rationalised that targeting the most primitive population of HSPCs that have the potential for long-term repopulation would ensure the greatest therapeutic benefit to patients, who need life-long WASp expression in HSPC cell lineages.
CRISPR offers several advantages over lentiviral gene therapy. It allows us to make one targeted integration of the wild-type WAS gene per cell tomaintain a normal cell copy number. We can also maintain proper transcriptional regulation by endogenous regulatory elements, ensuring physiologically relevant WAS expression upon integration of the cassette. This is essential to completely and reliably correct the disease phenotype in all affected haematopoietic lineages and to minimise the risk of persistent complications including autoimmunity and thrombocytopenia as observed in lentiviral gene therapy clinical trials.«
The team developed a CRISPR-Cas9 platform to knock-in a full-length WAS complementary DNA (cDNA) in frame with its endogenous translational start codon. This would allow transcriptional regulation from the WAS gene’s endogenous promoter and enhancers. The K562 human leukaemic cell line and HSPCs from healthy male donors were used to develop and optimise the editing strategy including the selection of the most efficient guide RNA (gRNA). Cas9 protein and gRNA were delivered to cells via preassembled ribonucleoprotein (RNP) complexes following electroporation.
To deliver the donor DNA molecule that would serve as a template for homologous recombination-mediated repair, the group created an adeno-associated virus type 6 (AAV6) vector that harbours a promoterless wild-type WAS cDNA with 800 bp-long upstream and downstream homologous flanking regions. The presence of a GFP reporter gene on the AAV6 vector allowed the researchers to calculate integration rates and after extensive optimisation of the editing protocol and culture conditions, the group achieved targeted integration frequencies of up 61 % in patient-derived HSPCs. Up-scaling of the optimised protocol using clinical-grade equipment yielded similar high integration rates without any negative impact on cell viability. Figure 1 shows a graphical representation of the workflow used to correct WAS.
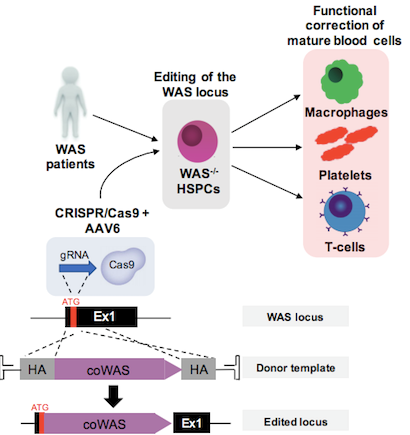
Insertion of a single copy of wild-type WAS cDNA in patient-derived HSPCs was sufficient to restore morphological and functional defects in downstream progeny i.e. mature haematopoietic cells including macrophages, platelets, T cells and B cells.
Initial experiments revealed that knock-in of a WAS cDNA with its own polyA tail could not recapitulate physiological WASp expression levels. To address this, the group developed a second cDNA construct that instead included a synthetic, commercially available mammalian terminator sequence (known as BGH), which is broadly used in vectors destined for gene therapy applications. This amended cDNA yielded wild-type WASp expression levels in all cell types tested across different experiments and independently of cell donor sources.
This work shows for the first time in human healthy and patient-derived HSPCs that it is possible to restore wild-type WASp expression levels by knocking-in one copy of WAS in its endogenous locus. It is also the first side-by-side comparison of CRISPR and the existing lentiviral-mediated WAS replacement approach for this disease. Furthermore, the researchers observed that transplantation of edited patient-derived long-term repopulating HSCs led to functional and stable restoration of WASp expression levels in bone marrow and white blood cell progenies in an immunodeficient mouse model (Fact Box 2) for up to 26 weeks.
According to Cavazza, the work shows that CRISPR could lead to comparable, if not better, results than lentiviral therapy.
»What makes our results unique compared to other gene editing approaches are the high rates of targeted integration in HSPCs and primitive stem cells as well as the high engraftment rates of edited patient-derived cells in immunodeficient mice that we were able to achieve, all in the absence of any relevant off-targeting effects.«
The most current lentiviral gene therapy for WAS is a product called OTL-103, which is undergoing clinical development by Orchard Therapeutics, UK. OTL-103 is made from patient-derived immature bone marrow cells and is recommended for the treatment of severe WAS cases, which represents only a portion of WAS patients. According to Cavazza, one of the drawbacks of this approach is that it doesn’t always reliably correct the platelet defects in treated patients.
The development of an autologous therapy that will reliably correct thrombocytopenia (low platelet count) would greatly benefit patients with milder forms of WAS in whom platelet defects are the main clinical consequence. This would have major beneficial impacts on bleeding risk and quality of life.
Collectively, WAS patients harbour about 300 distinct scattered mutations in the WAS gene, and the group’s gene editing strategy can correct all of these mutations.
Alessia Cavazza is hopeful that a CRISPR approach to WAS replacement may benefit more patients than lentiviral therapy: »Given the exciting preliminary results we obtained in an in vitro model of platelet differentiation from corrected human HSCs, we believe that our gene editing approach would be attractive to patients with classic as well as those with attenuated WAS, for whom allogeneic transplant and even lentiviral gene therapy presents unacceptable risks or the possibility of poor efficacy. This would extend the approach to a higher number of patients, and would likely several hundreds of additional patients in the UK alone.«
»We are working with humanised WAS mouse models to better define the curative potential of our strategy, and to determine what level of bone marrow engraftment and long-term persistence of edited cells is needed for full therapeutic benefit. So far, we have observed a correction rate of >70% in HSPCs and more primitive HSCs, and engraftment levels similar to lentivirally-transduced cells and unmanipulated WAS cells in NSG mice. These findings suggest that once we reach a sufficient engraftment level of edited stem cells, our strategy is likely to be curative, but we have many more experiments to perform yet.« said Alessia Cavazza.
The goal is to edit around 200 million haematopoietic stem cells per run, since this is in line with the upper limit of cells that would be transplanted into children if the therapy reaches the clinic.
The group is also addressing safety of its CRISPR WAS replacement construct and is planning to track genetically modified cells through time in vivo to monitor the genotoxicity potential of the treatment.
»If we are able to demonstrate efficacy and safety of the gene editing approach through late preclinical studies, we anticipate that our project will become attractive to major translational follow-on funding and industry engagement. We envision starting academically sponsored phase I/II clinical trials within a 3-5 year timeframe.« said Alessia Cavazza.
Link to original article in Nature Communications:
The NOD scid gamma (NSG) mouse is the current state-of-the-art model used to assess the rates of in vivo engraftment of human HSPCs into the bone marrow, and this model is widely used in gene therapy and gene editing pre-clinical studies for blood and immune diseases. Given the complexity of WAS, in vivo correction of its associated immunological deficiencies can be ultimately assessed only in WAS-/- mouse models. Two such models exist but it is not possible to transplant these mice with human cells because they are not immunodeficient and would therefore immediately reject the transplanted cells. Alessia Cavazza’s group is working to overcome this challenge by developing a humanised mouse model WAS-/- mouse model in which the murine WAS locus is replaced with the human WAS gene.
ArticleInterviewAdeno-associated virus (AAV)Lentivirus (LV)Severe Combined Immunodeficiency, SCIDWiskott-Aldrich Syndrome, WASGene therapyCRISPR-CasCas9SafetyInfection, Immunity & Inflammation Department at University College London





