FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Metachromatic leukodystrophy (MLD) is an incurable lysosomal storage disorder that can arise from any of approx. 300 known gene mutations in the ARSA gene. ARSA encodes the Arylsulfatase-A enzyme (ARSA), which plays a key role in sulfatide (a type of fat) metabolism in distinct cell types, including kidney and brain cells.
ARSA deficiency leads to perturbed sulfatide metabolism, causing increased sulfatide excretion in kidney cells without any associated pathology, a feature that is used in diagnostic testing for MLD. However, sulfatide accumulation in the lysosomes of cells from the peripheral and central nervous system results in progressive demyelination and neurodegeneration, with loss of motor function, language, and cognitive decline.
In a recent study published in the CRISPR Journal, scientists and medical doctors led by Dr Markus Mezger at the University Children’s Hospital at University of Tübingen, Germany, used CRISPR to integrate mutation-free ARSA cDNA into HSPCs from MLD patients. We recently interviewed several members of the team, who are very excited about the new findings:
»The results are promising: the edited cells display ARSA enzyme activity levels comparable to those seen in healthy individuals, and we are now working on advancing our findings as part of a stem cell gene therapy strategy for MLD,« said Dr Justin S. Antony, first author of the study.
Common treatment regimens for MLD include allogeneic HSPC transplantation or enzyme replacement, both of which are associated with low success rates. Allogeneic transplantation presents risks including graft-vs-host disease, the need for immunosuppression, and dependency on finding a healthy donor. Enzyme replacement therapy must be administered every one or two weeks intrathecally by lumbar puncture to cross the blood-brain barrier. Transplantation of lentiviral-modified stem cells that overexpress ARSA represents the most promising treatment so far, but only if applied before symptom onset.
MLD can be divided into three forms according to the onset of symptoms: early form (debuts before 2.5 years old), juvenile form (debuts from 2.5 – 16 years old), and an adult form. The age of onset and type of symptoms determine the course of the disease, which typically progresses most rapidly in the early forms.
When it comes to treatment, timing is of paramount importance, as Dr Krägeloh-Mann, who was also involved in the study, warns: »The treatments that are available until now have often not been successful, especially because treatment usually comes too late even if it starts in a pre-symptomatic stage. The cells involved become increasingly dysfunctional, which leads to a very devastating and progressive disease.«

Lentiviruses infect by inserting their DNA into the host cell genome, and they are widely used as delivery vectors in gene therapy to insert, modify, or delete genes in host cells.
Studies published in 2013 and 2016 reported that autologous HSPCs that were lentivirus-modified to overexpress ARSA prevented demyelination and led to increased ARSA enzyme activity in children with early onset forms of MLD. A clinical trial based on this approach was later sponsored by Orchard Therapeutics and resulted in marketing authorisation for Libmeldy™ for early-onset MLD by the European Medical Agency in late 2020.
However, despite the successes of lentiviral therapies, this delivery vector comes with safety concerns: more than 36,000 unique random and non-targeted genome integrations of ARSA-expressing lentivirus were found in one study. While no cancer development has been reported so far, such findings raise concerns about the potential for insertional mutagenesis.
The team therefore saw CRISPR as a natural alternative to lentiviral therapy for transgene integration, and the current study is the first to demonstrate the use of CRISPR in HSPCs to correct a central nervous system condition.
The team set out by designing more than 50 guide RNAs (gRNAs) that were predicted in silico to target the ARSA gene region. From these, 5 were eventually selected for experimental validation of the on-target cleavage potential against the ARSA UTR in a cell-free in vitro cutting assay, where each gRNA was complexed with the Cas9 ribonucleoprotein (gRNA/Cas9) and incubated with a PCR product containing the target region.
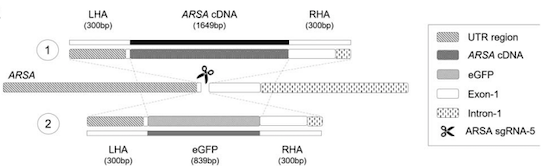
To deliver the cargo to cells, a gRNA/Cas9 complex and a repair template carrying a mutation-free, codon-optimised ARSA cDNA was cloned into the adeno-associated virus 6 (AAV6) vector. The team used a similar delivery method in a previous publication describing correction of mutations in the β-globin gene in β-thalassemia patient-derived cells.
Dr Mezger explains the rationale behind gene integration: »Gene correction is more challenging because ARSA is a big gene [around 3.2 kb] with multiple introns and exons and regulatory elements. It is quite challenging to circumvent the size limitation of AAV when we need to include homology arms and a poly-A sequence if we opt for a gene-correction strategy.«
In initial experiments, all 5 gRNAs showed a high level of on-target editing (88-97%). The team then validated each gRNA in HSPCs from a healthy donor, and the gRNA that resulted in most efficient gene editing and least detectable off-target editing was chosen for subsequent proof-of-concept studies on targeted transgene integration of ARSA cDNA in HSPCs.
Transgene integration was targeted to the endogenous ARSA locus under the control of the endogenous ARSA promoter, with the goal of achieving targeted integration and transgene expression at endogenous levels. The repair template further contains a poly-A tail after the transgene sequence to prevent transcription leakage of the mutated gene (Figure 1).
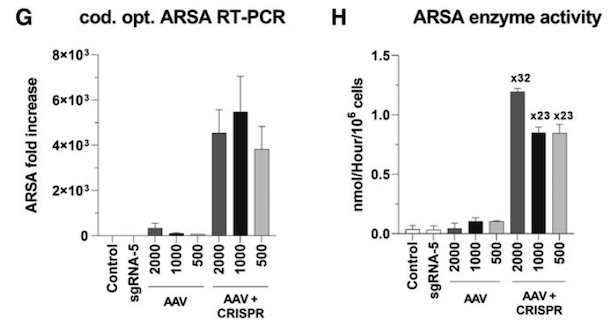
The gRNA/Cas9 complex and the AAV6 vector were introduced into healthy HSPCs via electroporation, which yielded a 32.7% transgene integration rate as measured by digital droplet PCR. This resulted in a >220-fold increase in mutation-free ARSA expression, as compared to untreated cells. Endogenous ARSA transcripts decreased by 60-70%, as determined by RT-PCR.
The design of the system allows one to easily assess transgene introduction and expression by PCR, because the transgene harbours a codon-optimised sequence, making it possible to distinguish it from the endogenous ARSA gene by specific amplification of each template.
With encouraging results from initial in vitro experiments showing that ARSA cDNA integration was possible, the team proceeded to perform proof-of-concept studies on HSPCs collected from two MLD patients carrying distinct ARSA mutations.
Using the exact same workflow as before, the team found that its CRISPR-based transgene integration system worked convincingly in patient-derived cells too.
In cells from the first patient, ARSA transcripts increased >3,000-fold and enzyme activity jumped 32x (which is similar to levels seen in healthy cells). In cells from the second patient, ARSA transcripts increased >800-fold and enzymatic assays using an artificial substrate revealed that ARSA enzyme activity increased 20x (Figure 2).
The integration of a mutation-free ARSA cDNA represents an elegant solution to potentially correct a disorder that can arise through hundreds of different mutations, as Dr Antony highlights: »We decided to go for a universal approach whereby the same treatment modality can be applied to any MLD patient, regardless of the mutation (s) present.«
On-demand available here.
A significant advantage for the clinical translation of the new methodology lies in the fact that it works with patient-derived or autologous HSPCs. Dr Rupert Handgretinger, paediatrician & expert in stem cell transplantation & immunotherapy, who was also involved involved in the study details: »When we perform an allogeneic transplant [using cells from a healthy donor], we normally have a backup of autologous cells from the patient in case the graft is rejected. Using the new methodology, the patients would receive their own cells, circumventing the risks of allogeneic transplantation.«
Going forward, the team will leverage its extensive expertise in isolating and handling stem cells towards developing a new therapy for MLD. Dr Peter Lang, acting medical director of the Deptartment of Haematology/Oncology at University Children’s Hospital Tübingen, details: »Autologous HSPCs can be isolated and purified, gene edited ex vivo with the ARSA-encoding AAV vector and transplanted back into the patients. Alternatively, the purified gene-edited stem cells can be cryopreserved and then thawed according to standard procedures. The autologous transplantation setting would be the same as we use for other diseases, such as β-thalassemia.«
While the results so far are promising, it should be noted that the use of CRISPR to edit stem cells often comes with a degree of cytotoxicity. Dr Antony reveals that HSPCs are sensitive to DNA strand break induction which tends to activate the p53 pathway, leading to apoptosis. He reveals strategies the team is now exploring to solve this problem: »One possibility is to edit more cells, so that although some are lost, there are enough cells to perform the engraftment. Another option that we are exploring is modulating the p53 pathway. We are looking into transiently inhibiting p53 activation as a means to enhance cell viability after gene editing.«
Dr Lang is confident that the new approach can be advantageous in the treatment of MLD at any stage, but until it reaches clinical trials and enough clinical data can be generated, every available treatment option should be considered: »I believe we will have a lifespan in which both approaches [allogeneic and gene-edited autologous transplantation] will exist, and we will get enough experience to decide which approach will be the best one. It would be very interesting to compare such a gene-editing autologous approach to the currently existing allogeneic approach.«
Even with such amazing breakthroughs in gene therapy approaches, Dr Mezger alerts for additional clinical difficulties, such as the need to create a niche for stem cell engraftment: »There is a need for conditioning before transplantation, and the use of chemotherapy to create this niche in which the edited stem cells can engraft is an important factor.«
Dr Handgretinger shares that the medical and research communities are working on ameliorating this niche prerequisite: »We are working on generating this niche without chemotherapy, possibly by immunological methods, which would not be possible in the allogeneic setting.«
Dr Krägeloh-Mann agrees with these assessments and reiterates that timing is a key factor: »It will be very important to start the treatment early. We believe that in the long run it will be a combination of stem cell transplantation with a gene therapy approach, together with intrathecal enzyme delivery during the first months until the engraftment is ready to produce sufficient enzyme dosage.«
The team is actively looking to further improve the system: »At the research level, we are looking to have viral-free repair templates without having to use AAV. We also intend to reduce the cytotoxicity associated with the gene-editing process and are developing quality control analytics to propel good clinical translation of the system«, says Dr Antony.
They are also working on leveraging the codon-optimised repair template to achieve supraphysiological enzyme expression. This is needed because brain diseases require a cross-correction approach: edited myeloid precursor cells migrate into the brain and secrete ARSA enzyme that will be assimilated by the deficient brain cells. This migration phenomenon is rare, and requires the migrated cells to express the transgene abundantly.
According to the team, their method or similar can be applied to other neuro-metabolic conditions. Dr Ingeborg Krägeloh-Mann details: »If it works in one neuro-metabolic disease, it should work in another that affects another stage of the metabolic pathway. It is an extraordinary perspective, especially since the approach is not dependent on specific mutations.«
The team is confident that its system can be provide a lasting solution for MLD patients. »The idea is not to have multiple transplantations; we believe that a single one is what it takes«, Dr Mezger says. They are also encouraged by ongoing clinical trials, such as Vertex Therapeutics’ and CRISPR Therapeutics’ ongoing trial for CTX001 in sickle cell disease: »This study has been running for about two years now and the data shows that CRISPR is working in humans«, Dr Handgretinger states.
The team wants to start preparing a clinical trial and believe the technology can be deployed soon with the help of an industrial partner. »This work can be rapidly finished within a year. Ideally, we could reach the clinic within one or two years«, Dr Antony reveals.
Link to the original article in the CRISPR Journal:
A Mutation-Agnostic Hematopoietic Stem Cell Gene Therapy for Metachromatic Leukodystrophy
Pedro Pais is a molecular biologist and microbiologist based in Portugal.
ArticleInterviewNewsViralAdeno-associated virus (AAV)Metachromatic leukodystrophy (MLD)CRISPR-CasCas9





