FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...
Intellia Therapeutics announced on the 6th October that the New Zealand Medicines and Medical Devices Safety Authority (MEDSAFE) had authorised its application to initiate a Phase 1/2 trial for NTLA-2002 in adults with hereditary angioedema (HAE). This is the first single-dose genome-editing candidate for HAE to enter a clinical study.
HAE is characterised by severe inflammatory attacks with swelling in various organs and tissues. Plasma kallikrein is a protein that is known to drive multiple inflammatory pathways, including the production of the inflammatory mediator bradykinin, which is overproduced in HAE. Plasma kallikrein inhibition is among the current treatment approaches to HAE (see Fact box).
NTLA-2002 is an in vivo CRISPR-Cas9 gene-editing therapy candidate that is designed to knock out the target gene kallikrein B1 (KLKB1) in hepatocytes. This gene encodes prekallikrein, a precursor of plasma kallikrein, thus its knockout permanently reduces plasma kallikrein activity and halts the production of bradykinin to prevent HAE attacks.
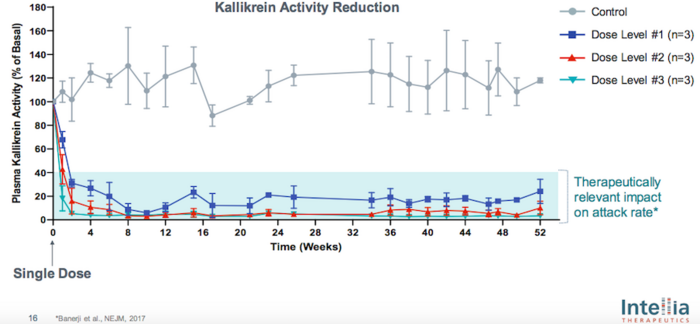
NTLA-2002 is delivered as a Cas9 mRNA and gRNA via lipid nanoparticles, and pre-clinical studies demonstrated sustained and therapeutically relevant reduction in plasma kallikrein activity following a single dose in non-human primates (Figure 1).
The new therapy will be given as a single dose, and according to a company press release, Intellia believes that NTLA-2002 has the potential to cure HAE and thus eliminate the current treatment burden associated with chronic therapy. NTLA-2002 is wholly-owned by Intellia Therapeutics, and the company expects to enroll the first patient by the end of 2021.
NTLA-2002 is Intellia Therapeutic’s second in vivo genome-editing candidate to enter clinical trials. Earlier this year, the company reported intermin positive Phase 1 data for NTLA-2001, which is being developed as a single-dose treatment for hereditary transthyretin (TTR) amyloidosis with polyneuropathy (ATTRv-PN).
The combined Phase 1/2 trial will evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of NTLA-2002 in adults with Type I or Type II HAE. This includes the measurement of kallikrein protein levels and activity as determined by HAE attack rate measures. The Phase 1 part of the study will follow an open-label, single-ascending dose design to identify up to two dose levels of NTLA-2002 that will be further evaluated in the randomised, placebo-controlled Phase 2 portion of the study. This Phase 1/2 study will identify the dose of NTLA-2002 for use in future clinical studies.
Intellia Therapeutics shared in its press release that it is currently working on submitting regulatory applications in countries beyond New Zealand.
We will provide further updates on the NTLA-2002 clinical trial as they become available.
For a complete overview of CRISPR IND approvals and ongoing gene-editing clinical trials, check out CRISPR Medicine News' Clinical Trials Database.
(HAE) is a rare, autosomal dominant disorder that is characterised by severe, recurring and unpredictable inflammatory attacks with swelling in organs and tissues throughout the body. Globally, approximately 1 in 50,000 to 1 in 150,000 people are estimated to be affected by HAE, and the most common disease form is HAE type I, accounting for about 85 % of all cases. This arises from a deficiency in the complement component C1 esterase inhibitor, which plays an important role in controlling inflammation. HEA type II is a more rare form of the disease, occurring in about 15 % of cases, and arises from abnormal C1 esterase proteins that do not function properly.
HAE attacks may be painful, debilitating and life-threatening. Current treatments include a number of approved replacement therapies using preparations of C1 esterase inhibitor to correct the deficiency, or drugs that inhibit plasma kallikrein, which in turn halts the production of the inflammatory mediator bradykinin to reduce pain and swelling as well as prevent HAE attacks. These treatments are life-long, and may require once- or twice-weekly intravenous or subcutaneous administration, or daily oral administration to ensure constant pathway suppression for disease control. Despite chronic administration, breakthrough attacks still occur, and the disease remains incurable.
ArticleNewsIn vivoLipid-based nanoparticleHereditary angioedema, HAEOther Genetic ConditionsRare DiseaseCRISPR-CasCas9Intellia Therapeutics, Inc.Clinical





