CRISPR-Armed Phages Clear First Human Safety Bar
CMN Interview
Mar. 3, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

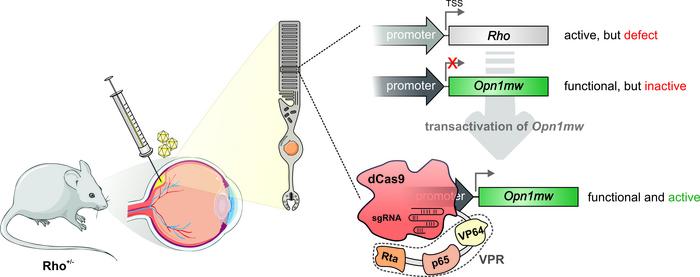
A novel and promising CRISPR-Cas gene therapy approach has been applied successfully for the first time in an animal model. By coupling transcriptional activators to 'dead' dCas9 researchers can target and activate a silent gene that can take over the functional role of a 'broken' mutated gene.
In the new proof of principle study the researchers successfully alleviated hereditary blindness in a mouse model of retinitis pigmentosa, a genetic disorder that affects millions of people worldwide.
»We are convinced that our approach could soon be used in first clinical trials,« says lead researcher Elvir Becirovic at the Chair of Pharmacology, Department of Pharmacy, Ludwig-Maximilians-Universität München, Germany.
Furthermore the approach could be applied to a range of other hereditary diseases. One particularly appealing aspect is that the activation approach avoids the risk of indel-type off-target effects in the genome because the DNA remains intact and unbroken.
The novel gene therapy approach takes advantage of the fact that many genes in our genome have similar functions. Often such functional equivalent genes are expressed in different cell types and tissues, and typically in cells where one gene is expressed, the other gene is silenced. But in principle, if the cells could somehow wake up the 'sleeping' gene, it could function as a back-up for a mutated gene.
»The central idea of our strategy was the assumption that switching on silenced, functionally equivalent genes in the affected cells can compensate for the missing function of the mutated gene and thus treat the disease,« says Elvir Becirovic.
Retinitis pigmentosa is the most common form of hereditary blindness, which affects millions of people worldwide and leads to a progressive and degenerative loss of photoreceptor cells. It primarily affects the rod photoreceptor cells in the retina that are responsible for night and dim light vision. One of the leading causes is mutations in the rhodopsin gene (RHO), which is only expressed in the rods. Depending on the genetic defect patients suffer from night blindness and gradual vision loss, which can eventually lead to complete blindness.
The other type of photoreceptor cells in the retina are called cones. These are responsible for colour and daylight vision, and they express different opsin genes that are functionally and structurally related to the rhodopsin gene. Many mammal species, including mice, express two opsin genes, while humans express three: OPN1MW, OPN1SW, and OPN1LW.
Just as the rhodopsin gene is silent in the cones, the cone opsin genes are silent in the rods. So the idea was to wake the cone opsin genes in the rods to see if they could replace the faulty rhodopsin protein, and alleviate blindness. And this is where the incredible 'DNA search engine' capabilities of CRISPR-Cas comes in.
Previous work has shown how the catalytically inactive 'dead' dCas9 can be used switch on silenced genes in the genome. The dCas9 is fused to transcriptional activators in complex called dCas9-VPR. The dCas9 is programmed as the classic CRISPR-Cas9 with a guide RNA that directs the complex close to the transcriptional start site of the target gene and the fused transcription activators call in the cell transcription apparatus to begin transcription.
To deliver the construct into cells, the researchers used the adeno-associated virus (AAV), whcih is the current 'gold standard' of delivery method within gene therapy. Since AAVs have a DNA packaging capacity limited to approximately 4.7 kb, the researchers used a dual AAV system that reconstitutes the split complex in vitro and in vivo in mice.
»There is no direct way to control that the dual AAV vectors will both target the very same cell. Still, we and others have already demonstrated that the so-called co-transduction efficiency of two separate AAV vectors is rather high. It is above 90% in the case of rod photoreceptors,« says Elvir Becirovic.
The researchers chose a mouse model for retinitis pigmentosa that carries a mutation in the rhodopsin gene.
The AAV vectors carrying the split construct were injected directly into the space between the photoreceptor cells and the retinal pigment epithelium.
AAVs will target all the different retinal cells, but to make the dCas9-VPR cell type-specific both parts of the dual AAV vectors were equipped with the rhodopsin promoter, whose expression is restricted to the rod photoreceptor cells.
Earlier in vitro tests had shown that both cone opsin genes could be targeted and activated with specific guide RNAs. However, for the in vivo mouse experiments, the researchers opted for Opn1mw gene, which they judged to be more suitable because of its spectral features and the expression pattern.
It all worked out as they had hoped and Becirovic and colleagues were able to activate the silenced cone opsin genes in the affected rods in a sustained and stable manner, and they followed the mice for up to 1 year after.
»This activation was able to compensate for the lack of rhodopsin function, slow down the degeneration process of retinitis pigmentosa and improve retinal function in this mouse model without detectable adverse effects,« says Elvir Becirovic.
An interesting strategy that the researchers did not look at in the current study would be to examine whether simultaneous activation of two or three of the opsin genes in mice and humans would improve the therapeutic success even more.
The proof-of-principle study is the first time the novel gene therapy approach using dCas9-VPR-mediated transcriptional activation has been shown to work for a genetic disease in an animal model.
Looking ahead, the strategy might be applicable to other genetic disorders as an appealing option over gene editing or gene supplementation therapy approaches, since many genes in the human genome are functionally equivalent.
»This approach can be extended to numerous other genes and hereditary diseases and offers key advantages compared to existing strategies,« says Elvir Becirovic.
He points to the carrying capacity of gene therapy vectors such as AAVs which limits gene supplementation strategies to small genes and says the novel activation approach »is independent of the size of the target gene.« In addition, Becirovic says the strategy in principle would allow therapies for more complex genetic diseases because it would be possible to switch on or off several genes simultaneously. Last but not least, the approach could be a safer strategy without the risk of unwanted off-target mutations.
»Due to the lack of enzymatic activity in the dCas9-VPR system, no off-targets at genome level are to be expected, as is a major concern with the classical CRISPR-Cas system,« says Elvir Becirovic.
Link to to the original article in Science Advances: A gene therapy for inherited blindness using dCas9-VPR–mediated transcriptional activation.
ArticleInterviewNewsDeliveryAdeno-associated virus (AAV)Hereditary BlindnessGene therapyTranscriptional activationOff-targetCRISPRa