
Aldevron Joins EGMEDC to Turn Genomic Promise Into...
May. 6, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Several CRISPR strategies have been applied to correct some of the thousands of documented mutations in the dystrophin gene (DMD) that cause Duchenne muscular dystrophy (DMD) - a fatal disease that leads to progressive muscle weakness and wasting. While most attempts have relied on the generation of DNA double-strand breaks (DSBs) by conventional Cas9 nuclease activity, only a few studies have taken advantage of base editing and prime editing. Since these relatively new methods only introduce single-strand nicks and make minimal change to the genome, they can serve as potentially safe and efficient alternatives to genetic correction of DMD disease-causing mutations.
The group of Eric Olson at the University of Texas Southwestern Medical Center in Dallas, Texas, followed this approach in developing a new CRISPR gene-editing strategy for DMD therapy that was published in Science Advances last Friday. The team addressed one of the most common disease-causing mutations, namely deletion of exon 51 (∆Ex51) in DMD, that disrupts the dystrophin reading frame and generates a premature stop codon in exon 52. The researchers managed to restore the reading frame with both base editing and prime editing by introducing exon skipping and reframing, respectively. In both cases, the strategy restored dystrophin expression in human cardiomyocytes, and contractile function was normalised using prime editing.
“This is the simplest possible way that we could imagine to restore the dystrophin protein by gene editing”Eric Olson
»The most important finding of this paper is that we can edit the dystrophin gene by modifying a single nucleotide - just one letter in the DNA code - and that is sufficient to cause skipping of a segment of the gene that results in restoration of the missing dystrophin protein. So this method offers a potentially safe and efficient way for correcting DMD in the simplest possible way,« says Eric Olson. He also points out that the approach can be adapted to target other mutations in DMD.
The Olson group has worked with Duchenne for several years, and in 2018 they published a review of the possibilities of using CRISPR in the therapy for DMD. His own work started with basic research on patient samples, and later, he moved to animal DMD disease models. Olson explains:
»In our initial studies, we used a form of CRISPR that cuts both strands of the DNA sequence. In the present paper, we further optimised and extended that correction procedure, so we don't have to cut both strands but only need to modify one letter in one strand to correct the disease. This is the simplest possible way that we could imagine to restore the dystrophin protein by gene editing.«
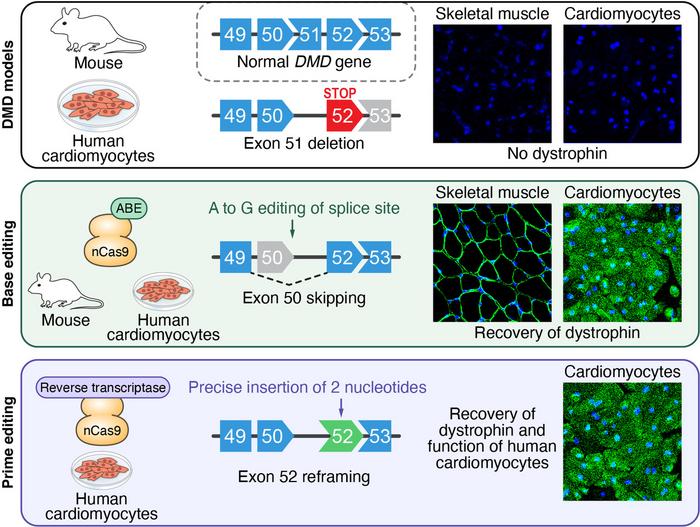
The team used both an animal and a human DMD disease model in their experimental work. The animal model employed mice with the ∆Ex51 mutation in their own mouse DMD gene. These mice displayed the hallmarks of DMD, e.g., absence of dystrophin and replacement of muscle fibres with inflammatory cells and fatty tissue. For the human model, Olson and his co-workers started with induced pluripotent stem cells (iPSCs) from a healthy male donor and used CRISPR-Cas9 to introduce the ∆Ex51 mutation in the human DMD gene. Edited cells were then isolated and differentiated into cardiomyocytes. Without subsequent experimental editing, differentiated ∆Ex51 cardiomyocytes lacked dystrophin expression, as demonstrated by immunohistochemistry.
In their first attempt to correct the mutated mouse DMD gene, the researchers targeted the splice acceptor site (SAS) or splice donor site (SDS) of either exon 50 or 52. Altering one of these splice sites will prevent the splicing machinery from recognising that exon so that it will be skipped, and the correct reading frame of the transcript will be restored. Since adenine base editors (ABEs) can convert an A:T base pair into a G:C base pair, they can destroy the canonical consensus sequences of both SAS and SDS, which are AG and GT, respectively.
»We developed this strategy called “single-swap” ABE, which means that the edit of just one letter in the splice acceptor or donor site can cause exon skipping and restore the correct reading frame of the dystrophin gene,« says Francesco Chemello. He is a postdoctoral researcher in the Olson lab and co-first author of the recent paper. The team used an optimised ABE, ABEmax, fused to an engineered Cas9 that can only nick one strand of the DNA, and then tested nine candidate sgRNAs for their ability to disrupt the acceptor or donor sites.
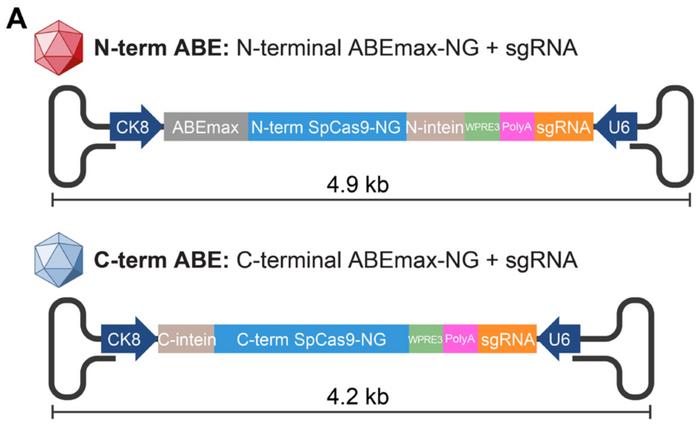
The team performed the initial tests in mouse N2a neuroblastoma cells. One of the sgRNAs induced 51% on-target editing in the SDS of exon 50 with less than 13% bystander activity. Bystander activity is editing of A:T base pairs adjacent to the on-target site. In this case one was located 2 nucleotides upstream and another one 4 nucleotides downstream of the on-target site. This sgRNA was then selected for in vivo gene editing in the mouse model using adeno-associated virus 9 (AAV9). The combined ABEmax and Cas9 construct, however, is too large to accommodate the 5 kb packaging limit of AAVs, explains Chemello:
»We needed to split the construct into two different AAVs, so the two partial proteins can recombine in the cell and form a functional base editor«. This was achieved by using the so-called split-intein packaging system previously established by David Liu at the Broad Institute of Harvard and MIT.
The single-swap gene-editing strategy and AAV split-intein delivery system was validated in the ∆Ex51 mouse model by intramuscular injections into the leg. Three weeks later, genomic DNA sequencing revealed on-target editing efficiency of 35%. Western blot analysis revealed that dystrophin protein expression had been restored to 54% of wild-type levels - up from around 5% in unedited ∆Ex51 cells. Moreover, immunohistochemistry revealed that dystrophin expression was restored in 96.5% of myofibers and inflammatory infiltration was markedly reduced.
“The preservation of normal muscle structure is restored after editing”Eric Olson
The team also tested the efficacy of the single-swap gene-editing strategy in a human disease model. Firstly, an sgRNA with 38% on-target editing at the SDS of exon 50 was selected using human HEK293T cells. Secondly, human ∆Ex51 iPSCs were nucleofected with this sgRNA and the base-editing machinery. Single clones of edited cells - 88% of which contained the on-target edit - were isolated and differentiated into cardiomyocytes.
DNA sequence analyses of these edited iPSC-derived cardiomyocytes revealed splicing of exon 49 to 52, indicating that the desired skipping of exon 50 had taken place. Like in the mouse model, the edited cardiomyocytes showed restoration of dystrophin protein expression as revealed by Western blot analysis and immunocytochemistry.
So the single-swap gene-editing strategy for exon 50 skipping works in both human and murine cells. And, as Eric Olson points out, the strategy also exerts functional, disease-alleviating changes in mouse muscles.
»That's an important point because, in this disease, the muscle tissue is destroyed and replaced by fibrotic scar and fat infiltration, so normal muscle structure is preserved after editing,” he says.
The promising in vivo results in the mouse model were obtained in muscle cells close to the injection site using an elevated viral dose. But one of the challenges with AAV is the high doses that are required to deliver expression throughout the body. So the Olson lab is already working to solve that problem.
»We believe, based on our preliminary studies, that we can eventually deliver this gene-editing machinery with a dose of AAV that is below the toxicity limit. But those are some of the issues that we are working on now,« says Eric Olson. This is partly achieved by guiding the AAVs to the cells that need correction.
»We only want to perform the correction where it is needed, that means in skeletal muscles and the heart, so we use this specific serotype AAV9 that mostly targets these two organs. Moreover, as a second layer of security, we use the creatine kinase 8 promoter that drives high-level expression specifically in skeletal muscle and heart,« Chemello explains. The postdoctoral researcher adds that the team will also need to use a smaller Cas9 so the whole construct can fit into a single AAV.
Olson, Chemello and the rest of the team then turned to prime editing for correcting the DMD causing ∆Ex51 mutation in human cardiomyocytes. This is the first time that prime editing has been used for DMD correction. In this case, the strategy was based on inserting two nucleotides into exon 52 to restore the reading frame. Prime editing utilises an engineered Cas9 that only nicks one strand of the DNA (nCas9) fused to a reverse transcriptase. A prime-editing guide RNA (pegRNA) is used to serve multiple purposes and consists of an sgRNA that anneals to the target site, a scaffold for the nCas9, a reverse transcription template containing the desired edit, and a primer binding site that binds to the non-target strand. In this case, an additional nicking sgRNA was included to increase editing efficiency by favouring DNA repair to replace the non-edited strand.
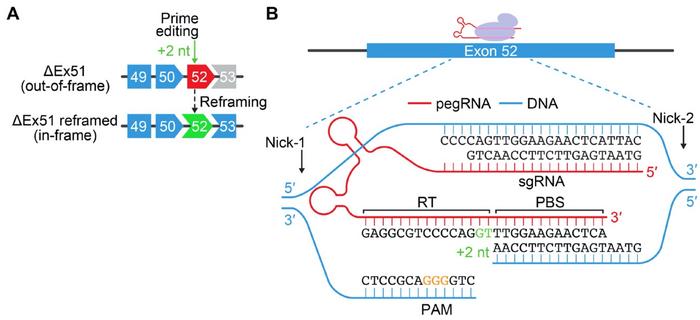
After a series of optimisation steps, the team selected two sets of pegRNAs and nicking sgRNAs that led to 20% and 54% efficiency in introducing a +2-nt insertion at the desired position in ∆Ex51 iPSCs. Single clones of edited cells were then differentiated into cardiomyocytes where correct reframing in exon 52 could be confirmed both by genetic analyses and by the restoration of dystrophin protein expression.
“This new research adds to the toolbox of genome editing technologies that we can deploy to correct genetic defects”Eric Olson
Moreover, the edited cardiomyocytes were subjected to calcium-cycling analyses to test if prime editing could rescue the arrhythmic defect seen in patient phenotypes. These experiments showed that 65% of unedited ∆Ex51 cardiomyocytes had arrhythmic calcium traces. This was, however, only the case in around 40% of prime-edited cardiomyocytes - a number very close to that obtained from healthy control cardiomyocytes of 37%.
»Our next step will be to demonstrate if this technology also works for other mutations in DMD. Our long term goal is to move this work to the clinic, but we don’t have a specific timeline established yet. We have to ensure safety and continue to optimise delivery methods,« says Olson. He concludes:
»This new research adds to the toolbox of genome-editing technologies that we can deploy to correct genetic defects. It may depend on the type of mutation as to which one of those technologies will be the most effective. We are more and more optimistic and hopeful about our path toward a new treatment for DMD.«
Link to the original article in Science Advances:
Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing
ArticleInterviewMost readNewsAdeno-associated virus (AAV)Duchenne Muscular Dystrophy, DMDGene therapyOff-targetBase editorsCRISPR-CasPrime editors





