Inhaled CRISPR-Cas9 nanoparticles disrupt Sting1...
CMN Briefs
Mar. 9, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...
Beta-thalassaemia affects tens of thousands of newborns each year and arises from mutations in the HBB gene that reduce or eliminate β-globin synthesis. The resulting imbalance with α-globin chains disrupts red-cell maturation and drives chronic anaemia, leaving many patients transfusion-dependent.
“The base editing mutation correction approaches described in this study change the therapeutic landscape for β-thalassemia both in terms of safety and efficacy”Giulia Hardouin
Existing gene therapies – lentiviral addition and CRISPR-Cas9 nuclease reactivation of fetal haemoglobin – have shown clinical benefit but retain drawbacks: variable efficacy in the most severe β⁰/β⁰ genotypes and concerns about double-strand breaks and their consequences. Giulia Hardouin at the Imagine Institute, Paris and first author of the paper that was published today in Science Translational Medicine, notes:
»By contrast with universal fetal haemoglobin (HbF) reactivation strategies, mutation-specific gene correction approaches could allow a more predictable and physiological production of higher levels of therapeutic haemoglobin, a determining factor for gene therapy efficacy in severe transfusion-dependent β-thalassemia (TDT).«
The study focuses on two prevalent β⁰ mutations – CD39 and IVS2-1 – that abolish β-globin production and are heavily represented among patients with severe disease. Using guides matched to these sites, the team targeted ABE8e variants to patient-derived haematopoietic stem and progenitor cells.
»In fact, we demonstrate that base editing usage allows for therapeutically relevant gene correction levels in patients’ hematopoietic stem cells carrying the most severe β0 mutations without relying on toxic double-strand breaks,« Giulia Hardouin notes, explaining the rationale succinctly.
Editing produced high correction rates – around 98% for CD39 and about 90% for IVS2-1 in differentiated progeny (see Figure 1). Restored HBB transcripts and β-globin protein were accompanied by a more balanced α/non-α globin ratio, reduced erythroid apoptosis, and improved maturation, including appropriate downregulation of early markers.
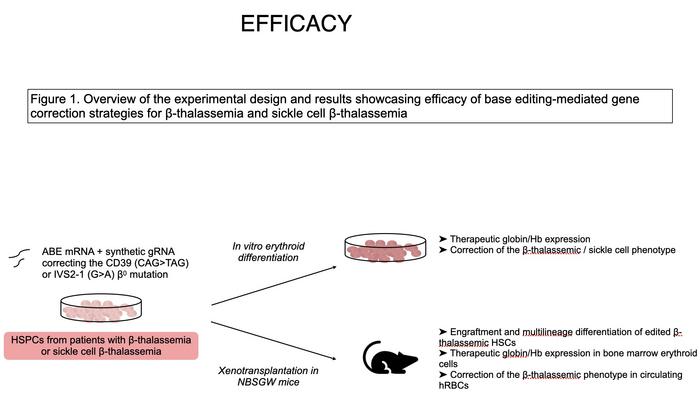
The approach was also applied to sickle-cell β-thalassaemia, where repairing the β-thalassaemic allele shifts cells toward the benign sickle-trait state. Deoxygenation assays showed a substantial reduction in sickling compared to untreated cells, with levels lower than in asymptomatic carriers.
»To our knowledge, our study describes for the first time the efficacy of such β-thalassemia mutation correction approaches to correct also the sickle phenotype associated with the sickle cell-β-thalassemia genotype, further expanding the scope of clinical application of these base editing strategies,« says Giulia Hardouin.
Safety was assessed across multiple complementary assays. GUIDE-seq and computational prediction identified candidate off-targets, which were validated in primary HSPCs. Structural integrity of the HBB locus was preserved after base editing, contrasting with deletion-rich alleles generated by Cas9 nuclease. Whole-exome sequencing in vitro and after xenograft engraftment showed no detectable increase in mutation burden, and clonal analyses indicated preserved stem-cell diversity.
»We used a combination of cutting-edge assays to confirm the safety of the highly processive ABE8e usage in patients’ HSPCs, in terms of genotoxicity and specificity,« Giulia Hardouin explains.
RNA-seq showed minimal transcriptome perturbation beyond the targeted correction, with no increase in A>G RNA deamination. Bystander edits – including benign Hb San Bruno and Hb-Nîmes – were functionally neutral in engineered HSPCs. A further screen identified an ABE variant capable of reducing C-bystander activity while maintaining high correction efficiency.
To gauge clinical reach, the authors examined the French NaThalY registry, Giulia Hardouin explains:
»Epidemiological studies are key to refining the population that could benefit from these approaches. Here, we took advantage of the French national thalassemia registry (NaThalY) and estimated that >40% of TDT patients who have not received an allogeneic HSC transplantation could benefit from such a treatment.«
The work was led by Annarita Miccio at the Imagine Institute (INSERM UMR1163) in Paris and conducted by a collaborative team of researchers at the Imagine Institute, the San Raffaele Telethon Institute for Gene Therapy in Milan, Genethon in Evry, and clinical partners in Marseille, with contributions from the University of Padova. It was published today, 26 November, in Science Translational Medicine.
To get more CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.
ArticleCMN HighlightsInterviewNewsBeta ThalassemiaBlood diseaseBase editors