CMN Weekly (20 February 2026) - Your Weekly CRISPR...
CMN Weekly
Feb. 20, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Decoding the genetic complexity of diseases often hinges on understanding how individual genetic variants affect protein function and drug response. For years, researchers have sought methods to explore the vast array of potential mutations in disease-associated genes. Yet, traditional approaches struggle to replicate the diversity and complexity of the human genome. A recent study takes a major step forward by employing advanced CRISPR-based technologies to map the functional impact of tens of thousands of mutations in the EGFR gene - a critical player in many cancers.
Led by Associate Professor Randall Platt at ETH Zürich, the study pairs cytosine and adenine base editors with prime editing to create a comprehensive framework for probing the genetic underpinnings of tumorigenesis and drug resistance. By combining these tools, the team identified not only well-known mutations but also previously uncharacterised variants that influence EGFR activity and sensitivity to tyrosine kinase inhibitors (TKIs). These findings allow for a more personalised approach to treatment by matching specific mutations with the most effective drug, addressing the needs of patients with rare mutations excluded from clinical trials.
The study's rationale was the explosion in the number of variants of uncertain significance (VUS) in genetic databases like ClinVar. Over the past decade, these variants have increased significantly, now making up about 50% of the entries in such databases. This growth is due to the broader adoption of genetic testing, especially in cancer diagnostics, where it is becoming routine to identify mutations driving the disease.
»We chose EGFR because it's a well-characterised oncogene, making it an excellent candidate for establishing our new technique. EGFR is routinely sequenced in cancer patients, providing a wealth of data, including many uncharacterised variants. It struck a balance between being well-studied and having a significant number of VUS to investigate,« says PhD Olivier Belli, who is a co-first author of the study published last month in Nature Biotechnology, sharing this role with Kyriaki Karava, a fellow member of Randall Platt’s group at ETH Zürich.
Professor Jakob Nilsson at The Novo Nordisk Foundation Center for Protein Research in Copenhagen also uses functional genomics to dissect cellular regulatory mechanisms and to obtain high-resolution information on protein function. He did not participate in the study but acknowledges the research:
»Belli et al. conducted a very careful study where they dissect EGFR genetic variants using ABE and CBE base editors and expanded this with prime editing. The study illustrates the power of base editing and prime editing technologies in providing single base resolution information which is fundamental to understand disease mechanisms.«
The experimental setup followed a classic CRISPR gene screen and combined two cutting-edge genome-editing technologies - base editing and prime editing - to investigate mutations in the EGFR gene. First, guide RNAs or pegRNAs were computationally designed and ordered as DNA oligonucleotides. These were cloned into plasmids, which were then used to produce lentiviral libraries. The lentiviruses, capable of integrating into the genome, were used to infect cells in culture.
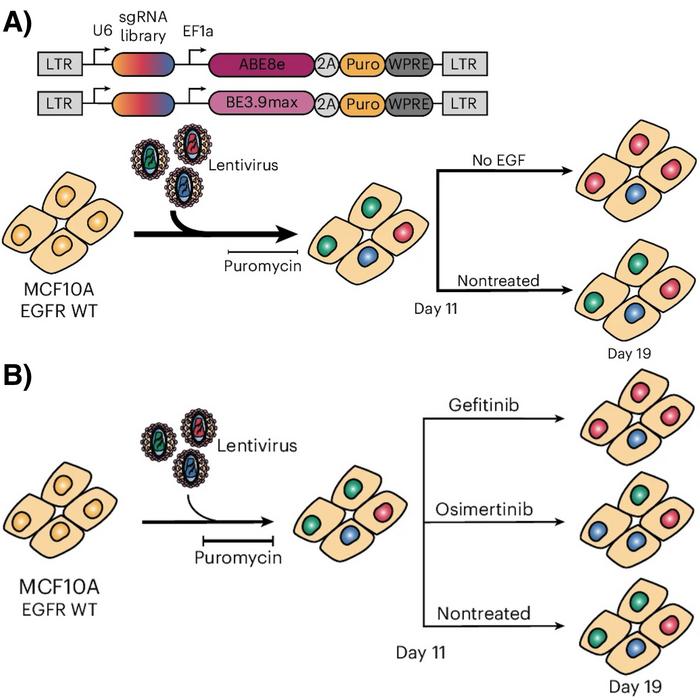
After selecting infected cells, each cell carried a single guide RNA or pegRNA, essentially acting as an independent experiment. Olivier Belli and his colleagues then subjected these cells to selective conditions, such as EGF deprivation or drug treatment. About a week later, they harvested the cells and used DNA sequencing to identify which guide RNAs were enriched or depleted, indicating their effect (see Figure 1).
The study used two complementary technologies: base editing and prime editing. Base editing allowed the scientists to perform high-throughput screens efficiently, introducing mutations across the gene. However, it needs more precision, often introducing multiple changes at once.
Prime editing, on the other hand, is highly precise and can introduce all types of mutations, including small insertions and deletions. This allowed the team to test variants observed in patients directly. Together, these methods provided comprehensive insights.
»For base editing, we used all possible guides targeting EGFR without filtering for known mutations. We then predicted the mutations each guide would introduce. With prime editing, we covered most of those identified in genetic tests,« Olivier Belli explains.
»In the future, we might use prime editing to target all possible bases in a gene systematically, but this would require massive libraries. Prime editing is currently less efficient than base editing, so we need redundancy - multiple pegRNAs targeting the same mutation to ensure success. That’s less of an issue with base editing, which works more consistently.«
Two assays, growth under EGF deprivation and drug treatment, were used to assess the functional impact of EGFR mutations. While effective for identifying whether a cell can proliferate in specific contexts, growth is a relatively rough readout that doesn’t capture the full range of phenotypic effects. Uncontrolled growth, a hallmark of cancer, makes these assays relevant for initial screens. Still, the results are seen as a baseline from which deeper analyses can explore how specific mutations influence receptor activation and downstream signalling pathways.
“The study provides important insight into EGFR mutations and cell proliferation as well as resistance mechanisms to tyrosine kinase inhibitors used in the clinic”Jakob Nilsson
A significant focus of the research was the ability to link specific mutations to observable phenotypes. While hits identified in the screens were validated as causal, non-hits posed challenges in interpretation, as their absence could result from inefficiencies in the editing process rather than an actual lack of impact. This uncertainty highlighted the need for further validation to understand the role of each mutation fully.
»The study is a technical tour de force that provides important insights into the power and limitations of base editing and prime editing technologies and shows the importance of validation. It provides important insight into EGFR mutations and cell proliferation as well as resistance mechanisms to tyrosine kinase inhibitors used in the clinic,« says Jakob Nilsson.
Olivier Belli also points out that there might be variants that influence growth under different conditions:
»For instance, EGFR is involved in both lung and brain cancers, but the mutations found in these two cancers often differ. In our screens, we identified mutations relevant to both cancer types. However, we only tested EGFR’s main ligand, EGF, even though EGFR has other minor ligands, such as amphiregulin. Some variants might only affect receptor behaviour in the presence of these other ligands. While we didn’t capture these effects in this study, the same approach could be replicated with other ligands to explore a broader spectrum of mutations.«
»There were many small “wow” moments when we discovered hits that hadn’t been found in patients before. Initially, I was sceptical, but digging into the literature revealed that many of these hits play roles in regulatory mechanisms. Even those not found in patients still taught us a lot about EGFR biology,« Olivier Belli explains.
One particularly unexpected discovery involved mutations in the receptor’s C-terminal tail, located on its intracellular side. These mutations disrupted splice sites, leading to a truncation of the C-terminal tail, a region critical for receptor signalling. This truncation caused constitutive activation of the receptor, resulting in some cases of drug resistance. While the involvement of the tail in regulatory mechanisms had been previously speculated, the specific mutation and its effects had not been characterised before this study.
These findings not only underscored the potential of prime and base editing to uncover novel regulatory mechanisms but also strengthened the case for deeper exploration of these pathways. By identifying mutations that disrupt key signalling and regulatory processes, the research provided new insights into receptor function and drug resistance, offering a foundation for further investigations into the underlying biology of these critical systems.
The importance of cellular context was a key takeaway. Two cell lines were used to study EGFR mutations: a wild-type line, MCF10A, and a cancerous PC-9 line with a pre-existing activating mutation commonly associated with cancer. The wild-type line served as a clean slate to study the isolated effects of single mutations.
In contrast, the PC-9 line modelled a more advanced cancer scenario where secondary mutations arise in a mutated background. This progression mimicked real-world cancer dynamics, where drug resistance often emerges through additional mutations. Interestingly, while some variants exhibited similar effects in both models, others conferred drug resistance only in one, emphasising the influence of the genetic and cellular environment.
»This highlights the importance of genetic and cellular context when studying variants. Expanding this study to include more cell lines with different primary mutations would undoubtedly yield further insights,« says Olivier Belli.
“Many EGFR inhibitors are already clinically approved, having gone through trials, but they’re not widely used because they’re not the most efficient for most patients. With our data, we could identify the most effective drug for specific patients without requiring new clinical trials since the drugs are already approved and available”Olivier Belli
A natural next step involves studying combinations of mutations, as many diseases result from interactions between multiple genes rather than isolated changes. While technically feasible with current tools, this approach is computationally and experimentally complex due to the exponential growth of possible mutation combinations.
A potential solution lies in testing single mutations or combinations directly in patient-derived cells, leveraging the relatively low toxicity of prime editing to preserve cell viability, Olivier Belli suggests:
»Approaches like prime editing are less toxic than classic CRISPR-Cas9, making them suitable for primary cells from patients. This would enable us to study mutations in their natural context, offering insights into personalised medicine.«
He sees prime editing essentially as a "dream tool" that is already showing promise but requires improvements in efficiency to realise its potential fully. Its versatility could expand to include larger deletions, gene inversions, and other complex edits that are biologically significant but currently challenging to reproduce. Such advancements would not only enhance the scope of studies like this but also pave the way for exploring the interplay of multiple mutations, reflecting the complexity of real-world diseases.
Olivier Belli emphasises that the study might help identify the most effective drugs for individual patients, especially among EGFR inhibitors that are already approved but underutilised. This approach avoids the need for new clinical trials and offers a straightforward path to personalised treatment by matching patients with the drugs most likely to work based on their unique genetic mutations. While the field is still developing standardised protocols and navigating regulatory hurdles, the ultimate vision is clear: fully personalised screens that quickly identify optimal treatments for cancer patients.
»Many EGFR inhibitors are already clinically approved, having gone through trials, but they’re not widely used because they’re not the most efficient for most patients. With our data, we could identify the most effective drug for specific patients without requiring new clinical trials since the drugs are already approved and available,« Olivier Belli explains.
After finishing his PhD at ETH Zürich, Olivier Belli has moved on from the lab, but the study has left a significant legacy. It establishes a robust and accessible protocol for exploring EGFR and potentially any other gene of interest. This framework empowers other research groups to apply the methods, even without extensive expertise in gene editing, to tackle their own questions about genes, mutations, or pathways.
By democratising the approach, the study has opened the door for further exploration into complex genetic interactions and their implications for disease and treatment.
Link to the original article in Nature Biotechnology:
Multimodal scanning of genetic variants with base and prime editing
To get more CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.
ArticleInterviewNewsLentivirus (LV)CancerCRISPR ScreensBase editorsPrime editors