CMN Weekly (13 February 2026) - Your Weekly CRISPR...
CMN Weekly
Feb. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Duchenne muscular dystrophy (DMD) is a severe progressive, degenerative disease that leads to muscle loss starting in early childhood, and patients have an average life expectancy of only 26 years. Several different mutations in the dystrophin gene (DMD) can cause DMD, and approximately 70% of the disease-causing mutations introduce frameshifts. Several CRISPR strategies have been used to create indels that delete the mutation or restore the reading frame. Since the dystrophin protein have been found to retain most of its function even with small internal deletions, this is an attractive strategy for DMD gene therapy.
In a recent paper, Yonglun Luo from Aarhus University in Denmark describes a new CRISPR gene-editing strategy for DMD therapy. The approach uses dual gRNAs to create precise and predictive DNA deletions, and Luo’s team demonstrates its capability to correct a DMD reading frame mutation in human fibroblasts. Moreover, the gene-edited fibroblast could be transdifferentiated into myotubes with restored dystrophin protein expression.
»I really like this study because it addresses a great need for treatment of Duchenne patients,« Luo says and goes on: »When I talk to neurologists at Aarhus University Hospital, they tell me that there is really a need for a cure because these patients are severely affected and die so early.«
Though many researchers have tried, it is not easy to efficiently and predictably introduce precise editing in DMD by CRISPR. So Luo decided to take advantage of a relatively newly discovered DNA repair mechanism. Indeed, it is so little known that it didn’t even have a name until he decided to call it non-homologous blunt-end joining (NHBEJ). And it is ideally suited for correcting frameshift mutations in DMD.
»For Duchenne therapy, this approach is actually ideal. NHBEJ offers an advantage to some CRISPR strategies that require high efficiency for predictive outcomes, and we demonstrate that it provides an efficient method to restore the dystrophin expression,« says Luo.
NHBEJ takes advantage of two gRNAs targeting each side of the intended deletion. After cutting by Cas9, the two double-strand breaks are repaired by blunt-end ligation without introducing any additional indels.
“Since we now can create a two-cut that will primarily repair by blunt-end joining, we are better able to control where and how long the deletion will be”Yonglun Luo
Luo and his co-workers demonstrated the reliability of NHBEJ using eight pairs of gRNAs targeting four human genes (TTR, CREB1, STAT2 and IRF9) and tested them in three different cell lines (HEK293T, HepG2 and HeLa). The deletion junctions were then characterised by both PCR, ICE (Inference of CRISPR Edits) and deep sequencing, and it turned out that NHBEJ accounted for 45% to 70% of all repair events.
»Since we now can create a two-cut that will primarily repair by blunt-end joining, we are better able to control where and how long the deletion will be,« Luo explains. This enables DMD gene therapy strategies that require either in-frame deletions that remove a disease-causing mutation or to make, e.g. +1 nucleotide deletions that restore the reading frame. He continues:
»In either case, you will lose a piece in the middle of the dystrophin protein, but even though it is not 100% identical to the wild-type, it is generally able to support 95% to 99% of the protein function. And that is enough to restore the muscle function of the patients.« The main reason for this is that DMD is the largest gene in the human genome and thus can allow for some missing parts. It spans about 2,300 kilobases, the transcript has 79 exons, and the final dystrophin protein contains 3,685 amino acids.
Luo is not sure why dual gRNAs promote NHBEJ. He explains that with one gRNA, the first cut of Cas9 would usually give rise to a 1 nucleotide insertion or deletion after repair. But subsequently, Cas9 can cut the repaired junction again since the target for the gRNA is only slightly altered. So the event keeps changing, and it takes some time before the indel profile stabilises. But this is not the case when you use two gRNAs, Luo explains:
»With dual gRNAs, you will delete a piece of DNA, and when they are put together, Cas9 cannot cut again.« Though this does not explain why NHBEJ occurs, it might lead to a more uniform indel profile. It also suggests that different repair mechanisms are operating when using either one or two gRNAs. Luo also points out that this is a very robust phenomenon:
»We haven’t really addressed the mechanism why this dual gRNA approach preferably leads to NHBEJ, but in our study, we measure nearly 20 pairs of gRNAs, and it is always the same. So it is not a random event.«
The team in Aarhus used the strategy to address a common cause of DMD that involves several missense or nonsense mutations in exon 51 of DMD. In-frame deletions of a small part of this exon can remove the critical mutation while retaining most of the protein function and potentially alleviate disease symptoms.
This experiment was performed using two pairs of gRNAs designed to generate in-frame deletions of either 180 bp and 168 bp if NHBEJ occurred as expected. Deletion junctions were analysed with ICE four days after plasmid transfection of HEK293T cells and revealed approximately 50% and 30% NHBEJ frequencies, respectively.
Luo and his co-workers then turned to human fibroblasts and another common cause of DMD. It is caused by deletion of exon 45, leading to a frameshift mutation and introduction of a premature stop codon in exon 46. This subsequently results in a truncated transcript that is degraded by the nonsense-mediated decay pathway.
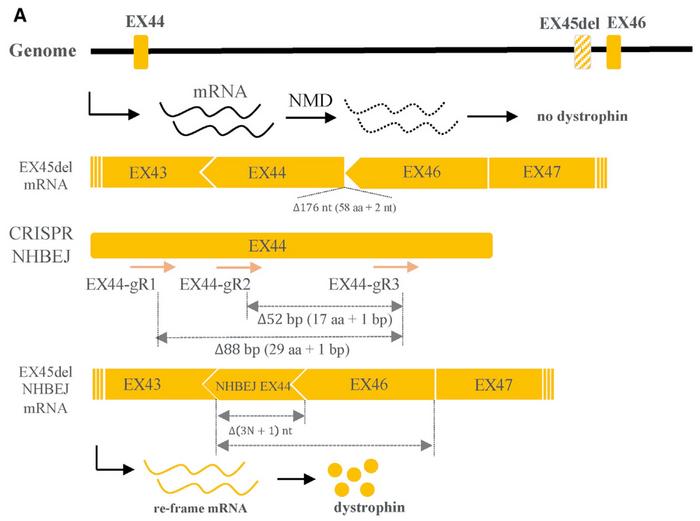
To correct this genotype, the team designed two pairs of gRNAs intended to introduce a 3n+1 bp deletion in exon 44. The gRNAs were designed, so the frameshift did not introduce a premature stop codon in exon 44 but rather corrected the reading frame in exon 46. After ribonucleoprotein delivery of the gRNAs and Cas9 into fibroblasts carrying the exon 45 deletion, NHBEJ repair accounted for more than 60% of all indels and restored the disease-causing mutation.
»To investigate if this approach could also rescue dystrophin expression, we developed a transdifferentiation strategy,« says Luo. Edited fibroblasts were transduced with lentivirus expressing human MyoD, which is a myogenic regulatory factor. After 12 days, the fibroblasts were transdifferentiated into multinucleated myotubes that expressed both dystrophin and myosin heavy chains, which are part of the thick filaments of skeletal muscle cells. Significantly, no dystrophin protein was expressed in un-edited cells carrying the exon 45 deletion mutation.
These results show proof of concept that dual gRNAs and NHBEJ can be used to efficiently correct mutations causing DMD in myotubes. For a therapeutic purpose, it would be ideal to use autologous muscle stem cells for correction and subsequent injection of edited muscle stem cells into the patient. But this approach is not practical, Luo explains:
»It is challenging to get muscle stem cells from Duchenne patients because they already have degeneration of muscle development, so the actual number of stem cells you can get from a patient is lower than a normal individual.« That is why Luo has decided to do the CRISPR gene editing on fibroblasts that are subsequently transdifferentiated.
“Our method can be used for all kinds of genes. For example, we could use it to remove the repeats or mutations causing Huntington’s disease, and we are looking into that now”Yonglun Luo
Going forward, however, the team needs to optimise the MyoD reprogramming step. So far, they have used lentiviral infection for this purpose, but it is not desirable in a clinical setting. Moreover, the process is inefficient, and only approximately 20% of fibroblasts are transdifferentiated. So Luo and his co-workers are working on a transfection method that does not rely on lentivirus and is both more efficient and compliant with Advanced Therapy Medicinal Products (ATMP) regulations.
Moreover, the researchers at Aarhus University plan to study the function of assembled myotubes in vitro after editing and transdifferentiation. They also intend to test their strategy’s performance in the X-linked muscular dystrophy (mdx) mouse model. But the dual gRNAs and NHBEJ strategy can do much more than correct Duchenne causing mutations.
»Our method can be used for all kinds of genes. For example, we could use it to remove the repeats or mutations causing Huntington’s disease, and we are looking into that now. Another exciting possibility is to investigate the function of a particular domain in a complex protein by precisely deleting it,« says Yonglun Luo.
Link to the original article in Molecular Therapy Nucleic Acids:
Efficient correction of Duchenne muscular dystrophy mutations by SpCas9 and dual gRNAs
ArticleNewsNon-viralLentivirus (LV)Duchenne Muscular Dystrophy, DMDCRISPR-CasCas9