FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Recently, the FDA placed a temporary hold on the ALPHA2 clinical trial conducted by Allogene Therapeutics after a chromosomal abnormality of unclear clinical significance was identified in TALEN-edited ALLO-501A CAR T-cells in a single patient.
Although further investigation is needed to determine if the TALEN gene-editing approach caused the observed karyotype abnormality, the fact that DNA double-strand breaks can pose a threat to genomic integrity in a cell is well known.
»Already in the first half of the previous century, Barbara McClintock showed that chromosome breakage can result in karyotype abnormalities,« begins David Pellman, professor of paediatric oncology and cell biology. His lab at Dana-Farber Cancer Institute focuses on genomic instability and applies state-of-the-art live cell imaging, single-cell analysis, and next-generation sequencing techniques to unravel the biological mechanisms that underlie chromosomal instability.
A causal correlation between chromosomal instability and malignant transformation has been firmly established ever since the first abnormal extrachromosomal bodies were identified in chronic myeloid leukaemia cells in 1960. »It's now known that whole genome duplication happens in something like 40% of all human cancers,« says Professor Pellman. Following his work on mitotic spindle formation in Saccharomyces cerevisiae, data from comprehensive human cancer genome sequencing projects triggered his interest in chromosomal instability, and more recently the genomic consequences of CRISPR gene editing.
When talking about CRISPR medicine risks in general, most concerns seem to be focused on potential off-target effects. However, various cellular processes — including incorrect repair of DNA double-strand breaks (DSBs) — may actually produce unwanted outcomes at the intended genomic target. To document these ‘unwanted on-target side effects’, Pellman’s lab has published several seminal studies. In a collaboration with the lab of Rudolph Jaenisch at MIT, they recently performed a comprehensive single-cell study for whole-genome sequencing of the early mouse embryo with and without CRISPR gene editing.
In that study, Cas9 protein, in combination with a guide (g)RNA targeting chromosome 2 or 17, was delivered into mouse zygotes by electroporation. Next, the development of these early embryos was followed for three rounds of cell division, up to the 8-cell stage.
By staining genomic DNA with the fluorescent marker DAPI, genomic instability initiated by DSB formation can be seen in the form of micronuclei, small bubbles containing genomic DNA outside the cell’s nucleus, or chromosome bridges containing stretched genomic DNA pulled by opposing sides like a tug of war towards the newly forming daughter cells (Figure 1).
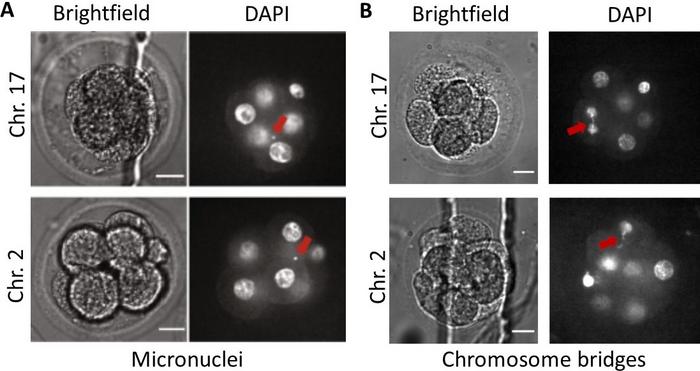
According to Professor Pellman: »A genomic DSB can cause the formation of a micronucleus or chromosome bridge when cellular DNA repair processes fail or produce particular errors. When a chromosome gets cleaved in two parts and the break is not repaired, a chopped off arm lacks the centromere structure for kinetochore attachment. During mitosis, this loose chromosome arm cannot segregate and it winds up being partitioned into a micronucleus.«
To explain the formation of chromosome bridges, David Pellman uses his hands: »Imagine my index finger is a chromosome and the tip is the Cas9 cut, right? So that's a chromosome in G1 of the cell cycle. After that's replicated, you get two side-by-side sister chromatids,« he aligns his two index fingers, »and both of them have unfinished ends due to the initial Cas9 DSB. The fact that those breaks are right next to each other means that they will ligate together!«
Professor Pellman then places the tips of his index fingers together and moves his hands in opposite directions and explains: »Since the sister chromatids each have a centromere connected by kinetochores to opposite poles, the DNA will be stretched out between two dividing cells and form a chromosome bridge.« The outcome of this chromosomal tug of war is variable, as the DNA bridge may break at a random location essentially resulting in an asymmetric distribution of chromosomal DNA between the daughter cells.
In their recent Nature Communication study entitled: ‘Whole chromosome loss and genomic instability in mouse embryos after CRISPR-Cas9 genome editing’, Pellman and colleagues explain that chromosome bridges can also cause whole chromosome loss: »Sometimes the entire chromosomal bridge ends up in one daughter cell, which also explains why the gain of a whole chromosome in one daughter cell results in the loss of a whole chromosome in the other,« says Pellman.
From bad to worse
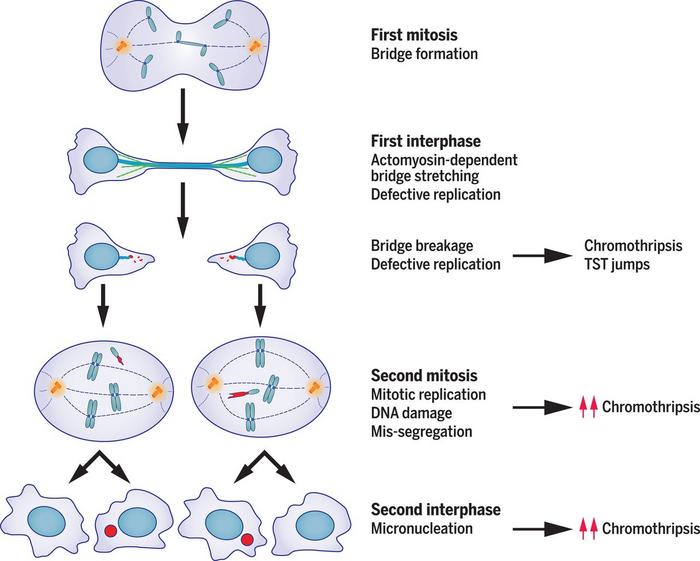
The observed mechanisms underlying chromosomal instability are not restricted to mouse embryos, and already there are indications that they might mean trouble for clinical gene editing in human somatic cells. »The formation of micronuclei or chromosome bridges is not where the cellular genomic instability ends, rather these abnormal DNA structures may snowball causing a cascade of localised genomic havoc,« continues Professor Pellman, who further elaborates: »Chromosomes in micronuclei undergo poor DNA replication. This under-replication really becomes problematic when the cell enters cell division and the nuclear envelope dissolves. The poorly replicated DNA fragments that arose in micronuclei are now free to reassemble with the cell’s chromosomes.«
Similarly, the asymmetric distribution of chromosomal DNA between the daughter cells following the shearing of a chromosomal bridge has consequences for the following cycle of DNA replication and cell division. »At the — now random — break site, there is no protection from a telomeric cap. Therefore, the genomic instability keeps snowballing because after DNA replication, the aligned sister chromatids will fuse again and form a new chromosome bridge or micronucleus during the subsequent round of mitosis.«
Micronuclei, chromosome bridges, and chromothripsis
This brings us back to the intriguing data from the cancer genome projects and the laboratory of Dr. Peter Campbell at the Sanger Institute. Professor Pellman clarifies: »For example, a characteristic case of chronic lymphocytic leukaemia, where all the chromosomes were normal…except for one which looked like somebody blew it up — all the little fragment bits — and stitched them back together in random order and orientation to make a novel chromosome. »To explain these shattered chromosomes in cancerous cells, Professor Pellman and co-workers stuck their necks out in a 2012 Nature publication providing indirect evidence that aberrant chromosome segregation between daughter cells did not just cause aneuploidy, but could also result in massive localised chromosomal rearrangements, a process referred to as chromothripsis.
»Back then, we didn’t have the technology to capture daughter cells and perform whole genome single cell sequencing on both of them. But over time, we succeeded in developing these methods and showed that by making a micronucleus, we could essentially recapitulate all known sequence features of chromothripsis. And then, in a 2020 Science paper, we showed that a rather similar series of catastrophic genomic events can result starting from a chromosome bridge (Figure 2).«
To obtain accurate data on the true impact of chromothripsis in malignant transformation, Professor Pellman collaborated with Dr. Peter Park in the Department of Biomedical Informatics at Harvard Medical School who analysed whole genome sequences of 2,658 tumors and found that this shattering chromosome phenomenon is an important driver of genome evolution in human cancer, with frequencies of over 50% in several cancer types.
But why do cells with a persistent or misrepaired CRISPR-Cas9-induced double-strand break complete their cell cycle instead of dying by apoptosis?
Professor Pellman shares his thoughts: »In human cells, you need somewhere between 20 and 30 unrepaired breaks to activate enough of a cell cycle checkpoint that allows DNA repair. So, a CRISPR-Cas9 break may therefore fly under the radar of DNA damage checkpoints. Single unrepaired breaks will also slip past mitotic checkpoints, enabling cell division. Although the literature is not 100% in agreement, it is my take that the spindle assembly checkpoint is not robustly activated unless you actually damage the centromere directly. Thus, what may happen is that a single CRISPR-Cas9 break results in the formation of these chromosome bridges or micronuclei that can generate hundreds of DNA breaks in the next generation.«
»What it comes down to is that the real culprit here is the Cas9-induced double-strand break that remains unrepaired and results in aberrant DNA structures that may cause chromotripsis. Consequently, the application of CRISPR medicines that do not cause double-strand breaks, like base editing or prime editing, could potentially avoid or significantly decrease chromothrypsis risks. That being said, both these alternative gene-editing methods do create a single-strand DNA break, and we know that these can sometimes be converted into double-strand breaks during DNA replication. As such, these alternative gene-editing techniques, although a significant improvement, may not be fully bullet-proof either,« says Professor Pellman.
He continuous: »Another viable option may be to stop cells from going through the cell cycle when gene editing takes place. It's kind of an imperfect strategy as it has its own drawbacks, but in principle, you could arrest the cells in a stage where the editing would occur for a long enough period of time so that you allow the double-strand break repair to be complete. Then, the edited cells would never go down the path of forming micronuclei or chromosome bridges. Similarly, it should be possible to use protein tags, called ‘degrons’, that can target Cas9 for degradation depending on the cell cycle phase. A degron-Cas9 would only be active in G1, empowering repair of the double-strand break later in the cell cycle.«
Have you observed that CRISPR-Cas9 gene-editing can result in chromothrypsis? »In our analysis of gene-editing consequences in mouse embryos, we didn’t see chromothrypsis,« answers Pellman, who continues: »But, that didn't really surprise us considering the small sample size. Performing complete 20 x genome coverage sequencing of single cells is extremely expensive. So, we are limited in the number of cells we can analyse. We actually did analyse over 200 embryonic cells, and while we didn’t detect chromothrypsis, we clearly did see a 2- to 3-fold increase in the formation of micronuclei after Cas9 treatment.«
And chromosome bridges? »Using confocal microscopy, we indeed observed chromosome bridges in Cas9-edited embryos [Figure 1B]. But the small sample size and obscured visual perception in 3D embryos is preventing us from giving an exact frequency. Nevertheless, we can now explain whole chromosome loss by a bridge-mediated mechanism after genome editing in embryos, and we should not negate the snowballing effect of micronuclei and chromosome bridges through cell generations.«
In a recent Nature Genetics paper, Pellman’s lab in collaboration with Dr. Mitchell Weiss from St. Jude Children’s Research Hospital roughly mimicked a clinical protocol for inactivating the BCL11 enhancer as used in experimental CRISPR medicine for sickle cell anaemia.
»In a model cell system, we were able to see DNA sequence signatures of chromotripsis in ‘granddaughter cells’. We could also show that targeting of the BCL11 enhancer resulted in the formation of micronuclei in about 2.5% of human haematopoietic stem cells. But, only a very limited number of these cells will possibly remain viable or even proceed towards chromothripsis,« states Professor Pellman.
»So there you have it,« concludes Professor Pellman. »At this time, we do not have a definite answer about the incidence rates of whole chromosome gains or losses, micronuclei or chromosome bridge formation, or chromotripsis events triggered by CRISPR-Cas9 gene editing. However, we have gathered sufficient evidence that gene editing-induced double-strand breaks have the potential to create aberrant chromosomal structures that have a causal role in malignant transformation.«
»Notably, CRISPR medicine in a clinical trial aiming to cure Leber Congenital Amaurosis blindness by injection of gene-editing tools in the eye is targeted to non-dividing cells. Moreover, haematopoietic stem cells at the center of several clinical CRISPR interventions are quiescent by nature. In these clinical settings, gene editing will likely take place outside the cell cycle stages that are causal to the formation of micronuclei or chromosome bridges.«
»As a physician who's taking care of both paediatric cancer patients and patients with diseases like sickle cell anaemia, I'm tremendously excited about CRISPR in general. But, at this time we just don’t know yet what the clinical risks are. We aim to learn from clinical trials. For example, when you're gene editing cells in a clinical protocol for ex vivo treatments like CAR T therapy, many millions of gene-edited cells are transplanted into the patient. Then it becomes a game of numbers! Obviously, there are strict monitoring and quality control procedures in place, but to get a true handle on the rate of CRISPR-Cas9-induced chromosomal insults, the CRISPR community really needs to come together. To perform the costly in-depth and comprehensive analyses required, we need to sift through thousands of single cells from sequential generations and test strategies to avoid these gene-editing risks. Only then can we safely unleash the full potential and benefits of this incredibly powerful and promising technology.«
Henri van de Vrugt is a molecular biologist and Chief Scientific Officer at New Haven Biosciences Consulting, US, and Senior Scientist & Advisor at the Dept. of Human Genetics, Amsterdam UMC, the Netherlands.
ArticleInsightInterviewMost readNewsIn vivoElectroporationCancerCAR-TCRISPR-CasCas9SafetyAllogene Therapeutics, Inc.





