CRISPR editors gain precision via evolved hotspots
CMN Briefs
Jul. 8, 2025


In September 2022, U.S.-based CRISPR company Excision BioTherapeutics announced in a press release that the first participant had been dosed in the Phase 1/2 clinical trial of EBT-101 for human immunodeficiency virus type 1 (HIV-1). According to that press release, the individual was dosed in July 2022, and initial safety findings indicate that EBT-101 has been well tolerated so far. The company also revealed that the participant continues to be monitored for safety and is anticipated to qualify for analytical treatment interruption of their background anti-retroviral therapy in an evaluation of a potential cure.
EBT-101 is the first CRISPR-based therapy to enter a clinical trial for HIV. HIV is an RNA virus, and when it first infects an individual, its RNA is reverse-transcribed into DNA by virally-encoded reverse transcriptase and RNase H enzymes that convert the viral RNA to double-stranded DNA (dsDNA) and degrade any remaining viral RNA, respectively. The resulting dsDNA is integrated into the host genome and this is referred to as proviral RNA.
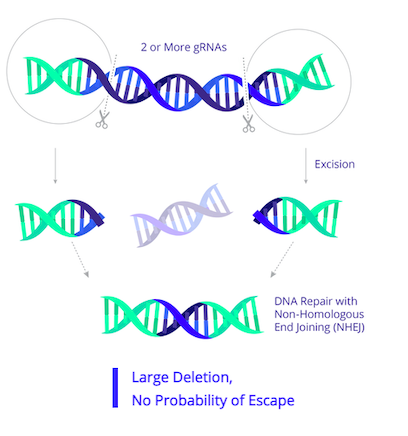
EBT-101 is designed to excise HIV proviral DNA using CRISPR-Cas9 and two guide RNAs (gRNAs). The CRISPR cargo is delivered to cells as a one-time treatment via an adeno-associated virus (AAV). The two gRNAs target three sites within the HIV genome (Figure 1), thereby cutting out large portions of the HIV genome and minimising potential viral escape.
On November 2nd 2022, Excision BioTherapeutics will host a key opinion leader webinar about HIV and the ongoing EBT-101 as a potential cure for HIV. Read more about EBT-101 in our previous clinical update.
Iovance Biotherapeutics announced earlier this month that the first patient had been dosed and completed the safety observation period in the Phase 1/2 IOV-GM1-201 trial for IOV-4001.
IOV-4001 is Iovance’s TALEN-modified, PD-1-inactivated tumour-infiltrating lymphocyte (TIL) therapeutic candidate for several solid cancers. IOV-GM1-201 is a first-in-human study investigating the safety and efficacy of IOV-4001 in patients with previously treated metastatic non-small cell lung cancer (NSCLC) or advanced melanoma.
IOV-4001 is developed using Cellectis' TALEN® gene-editing technology under licence. Specifically, it is generated from TILs that are isolated from patient tumours. TILs encompass a population of lymphocytes (including B cells, T cells, and other immune cell types) that naturally invade tumour tissue and are implicated in killing tumour cells. Following isolation, the TILs are subjected to TALEN gene editing to inactivate the PD-1 protein. IOV-4001 is designed as an adoptive cell therapy that combines the natural anti-tumour activity of TILs with PD-1 knockout to prevent immune escape by cancer cells.
PD-1 is an immune checkpoint protein found on the surfaces of healthy T cells as well as certain cancer cell types. Under normal physiological conditions, the interaction between PD-1 and its ligand PD-L1 mediates T cell immune suppression, but cancer cells can hijack this pathway to escape the immune response. The PD-1/PD-L1 interaction is a well-established target for cancer immunotherapy and to date various small molecule and antibody-based inhibitors of both interaction partners have been pursued as cancer treatments.
Caribou Biosciences announced last week in a press release that it had shared a case report on long-term follow-up data for the first patient dosed in its Phase 1 ANTLER trial at the Lymphoma, Leukemia, & Myeloma Congress 2022, which was held last week in New York.
CB-010 is the first allogeneic (i.e. donor-derived) CAR-T cell therapeutic candidate with a CRISPR-mediated PD-1 deletion to be cleared for a clinical trial. The candidate is currently being evaluated in the Phase 1 ANTLER trial for CB-010 in relapsed or refractory aggressive B cell non-Hodgkin lymphoma (r/r B-NHL).
The new data reveals that the first patient enrolled in the ANTLER trial had no detectable signs of B-NHL 15 months after receiving a single dose of CB-010. According to the press release, the patient in question was treated with dose level 1 (corresponding to 40x106 CAR-T cells). The patient, who is a 66-year-old male, was diagnosed in 2013 with aggressive follicular lymphoma, demonstrating disease progression within 24 months. Before joining the ANTLER trial, the patient had received eight prior lines of systemic anti-cancer therapy. After a single dose of CB-010, the patient initially demonstrated a complete response at 28 days, which has been maintained through his 15-month evaluation.
CB-010 is Caribou’s most advanced cell therapy candidate and is derived from healthy donor T cells that are genome-edited using the company's next-generation CRISPR-Cas technology that is based on CRISPR hybrid RNA-DNA (chRDNAs). Specfically, a CD19-specific CAR is inserted into the TRAC gene, which encodes the T cell receptor alpha constant. The PD-1 gene is also deleted in these cells. This gene encodes the PD-1 protein that functions as a safety switch on T cells that cancer cells turn on to protect themselves from T cell-mediated immune responses. CB-010 is the first allogeneic CAR-T cell therapy with a CRISPR-mediated PD-1 deletion to be cleared for a clinical trial.
Read about CB-010 and earlier clinical data in our previous clinical trial update here.
According to the U.S. National Cancer Institute (NIH), the overall response rate (ORR) is the percentage of individuals in a study or treatment group that exhibit a partial (PR) or complete response (CR) to the treatment within a certain period of time. CR specifically refers to the disappearance of all signs of cancer in response to treatment (also known as remission). Source: National Cancer Institute.
Allogene Therapeutics announced in a press release published last week that it has initiated a potentially pivotal Phase 2 clinical trial of ALLO-501A in patients with relapsed or refractory large B-cell lymphoma (r/r LBCL).
The trial, which is referred to as the ALPHA2 trial, is a single-arm Phase 2 study in r/r LBCL will utilise a single dose of ALLO-501A at 120 million CAR+ cells alongside a specific lymphodepletion regimen. The trial will enroll approximately 100 patients who have received at least two prior lines of therapy and have not received prior anti-CD19 therapy. The primary endpoint is objective response rate.
ALLO-501A is an anti-CD19 CAR T-cell therapy that is made from healthy donor cells using TALEN-based gene editing, and the company expects to provide an update on the programme toward the end of 2022.
In June 2022, the FDA granted RMAT designation to ALLO-501A in r/r LBCL. This designation was based on data demonstrating the potential of ALLO-501A to address an unmet need for patients with r/r disease. Read more about ALLO-501A in a previous CMN clinical trial update here.
Locus Biosciences announced last month that the ELIMINATE trial has begun enrolling patients, and that the first patient has been treated. ELIMINATE is a registration-enabling Phase 2/3 clinical trial for Locus' lead candidate LBP-EC01, a CRISPR-enhanced bacteriophage precision medicine designed to treat urinary tract infections caused by E. coli.
LBP-EC01 is a bacteriophage cocktail that is engineered with a CRISPR-Cas3 construct to target the E. coli genome. The precision medicine product works through a unique dual mechanism of action utilising both the natural lytic activity of the bacteriophage and the DNA-targeting activity of CRISPR-Cas3. In 2021, Locus reported in a press release that LBP-EC01 met all primary and secondary endpoints and demonstrated safety and tolerability in a Phase 1b trial.
Read more about Locus' approach and the Phase 1 trial for LBP-ECO1 in our interview with Locus Bioscience's CSO Dave Ousterout here.
Last month at the 2022 Bradykinin Symposium held in Berlin, Intellia Therapeutics shared positive interim clinical data for its second in vivo investigational CRISPR-Cas9 candidate, NTLA-2002, for the treatment of hereditary angioedema (HAE). HAE a rare genetic disease that is characterised by severe, recurring and unpredictable inflammatory attacks with swelling in organs and tissues throughout the body.
NTLA-2002 is designed to knock out the kallikrein B1-encoding gene in hepatocytes to permanently reduce plasma kallikrein activity and hence halt the production of the inflammatory mediator bradykinin, which is dependent upon plasma kallikrein, and which is overproduced in HAE.
The data presented are from the initial six adult patients with HAE in the ongoing dose-escalation study with a data cut-off date of July 27th, 2022. These participants received single doses of 25 mg (n=3) and 75 mg (n=3) of NTLA-2002 via intravenous infusion, and changes from baseline values of plasma kallikrein protein were measured for each patient.
The company revealed that administration of NTLA-2002 led to dose-dependent reductions in plasma kallikrein and achieved maximal reductions by week eight, with mean reductions of 65% and 92% in the 25 mg and 75 mg dose cohorts, respectively. Furthermore, these reductions were sustained through at least 16 weeks in the 25 mg cohort and eight weeks in the 75 mg cohort for which complete cohort biomarker data were available.
In addition to plasma kallikrein levels, HAE attack rates are also being measured in the ongoing study, and so far treatment at the lower dose level of 25 mg has led to a significant reduction or complete elimination of HAE attacks. At the time of the data release, patients on the higher dose level of 75 mg had not completed the primary 16-week observation period for HAE attack rate. Intellia expects to present HAE attack data for the 75 mg cohort at the American College of Allergy, Asthma & Immunology Annual Scientific Meeting in November 2022.
Intellia also announced last month that the FDA has granted the company with orphan drug designation for NTLA-2002 for treating HAE. Read more about HAE and the mechanism of NTLA-2002 in our previous clinical trial update here.
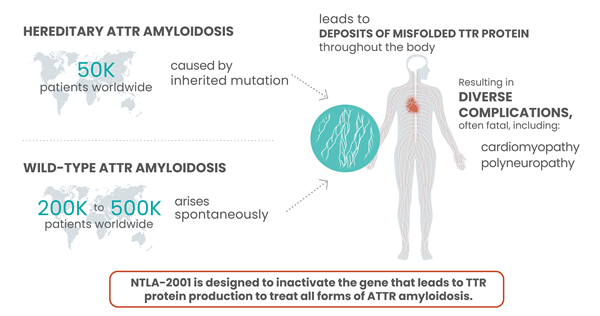
Intellia Therapeutics and Regeneron shared a press release last month announcing initial data from the cardiomyopathy arm of the ongoing Phase 1 trial of NTLA-2001, an investigational single-dose in vivo CRISPR-Cas9 therapy for the treatment of transthyretin (ATTR) amyloidosis.
According to that press release, the interim data include 12 adult patients with ATTR amyloidosis with cardiomyopathy (ATTR-CM) with New York Heart Association (NYHA) Class I – III heart failure. Single doses of 0.7 mg/kg and 1.0 mg/kg of NTLA-2001 were administered intravenously, and the change from baseline in serum transthyretin (TTR) protein concentration was measured for each patient. The data revealed that treatment with NTLA-2001 led to rapid and deep reductions of up to 94 % in serum TTR by day 28. In February 2022, the companies reported clinical data that revealed rapid, deep and sustained responses in a cohort of 15 patients with hereditary transthyretin (TTR) amyloidosis with polyneuropathy (ATTRv-PN).
ATTR is a rare, progressive disease, in which a protein known as TTR becomes misfolded and accumulates as plaques in tissues throughout the body. This causes serious complications that mainly involve the heart and nerves, and most patients die 2-15 years after disease onset. NTLA-2001 was the first in vivo CRISPR therapy to be administered to humans via the bloodstream. It is designed to treat ATTR by selectively reducing the levels of mutated TTR protein in the blood, through CRISPR-based inactivation of the TTR gene in liver cells.
Read more about the available clinical data for NTLA-2001 in a previous CMN clinical trial update here.
RMAT designation for T-cell cancer candidate CTX130
CRISPR Therapeutics reported in September 2022 that the FDA had granted a regenerative medicine advanced therapy (RMAT) designation to CTX130, for the treatment of cutaneous T-cell lymphomas. CTX130 is a donor-derived CRISPR-Cas9-edited CAR-T investigational therapy targeting CD70, an antigen expressed on various solid tumours and hematologic malignancies. The candidate is currently being assessed in two Phase 1 clinical trials: the COBALT-LYM trial for T or B cell cancers and a trial for safety and efficacy of several dose levels in renal cell carcinoma.
At the European Hematology Association Hybrid Congress in June this year, the company presented positive clinical results from the ongoing COBALT-LYM trial. CTX130 has previously received Orphan Drug Designation from the FDA for the treatment of T-cell lymphoma. You can read about the clinical data for CTX130 and its mechanism of action here.
First patient dosed in Chinese CRISPR trial for B-cell acute lymphoblastic leukaemia
Clinical-stage CAR-T company Gracell Biotechnologies (China) announced last week in a press release that it has dosed the first patient in its Phase 2 portion of a registrational Phase 1/2 clinical trial to evaluate GC007g for the treatment of B-cell acute lymphoblastic leukaemia (B-ALL). The trial is currently ongoing in China. GC007g is Gracell's allogeneic, human leukocyte antigen (HLA)-matched, donor-derived, CD19-directed CAR-T cell therapy candidate under development for the treatment of a subset of B-ALL patients who relapsed after allogeneic human stem cell transplant.
According to the same press release, data from the Phase 1 trial of GC007g showed encouraging efficacy and a favorable safety profile. The ongoing registrational Phase 2 trial will further assess the safety and efficacy of GC007g in r/r B-ALL patients at the recommended Phase 2 dose.
FDA grants a rolling review for sickle cell disease and beta thalassemia candidate exa-cel
CRISPR Therapeutics and Vertex Pharmaceuticals recently announced that the FDA has granted exa-cel a rolling review. Exa-cel (formerly known as CTX001) is an ex vivo CRISPR-Cas9-based candidate therapy that edits a patient's haematopoietic stem cells to reactivate the production of foetal haemoglobin (HbF). HbF is highly expressed and critical during foetal development, and is then rapidly suppressed early in life. HbF reactivation has emerged as an attractive strategy to treat sickle cell disease and beta thalassemia by compensating for the lack of functional adult haemoglobin seen in these blood disorders.
According to the FDA, a rolling review means that a drug developmemt company can submit completed sections of its Biologic License Application (BLA) or New Drug Application (NDA) for review by FDA, rather than waiting until every section of the NDA is completed before the entire application can be reviewed. Exa-cel is the most advanced clinical-stage gene-editing candidate for SCD and beta thalassemia to date. You can read more about the therapeutic strategy behind exa-cel and previous clinical data here.
For a complete overview of current gene editing clinical trials, check out CRISPR Medicine News' Clinical Trials Database.
To get more of the CRISPR Medicine News delivered to your inbox, sign up to the free weekly CMN Newsletter here.