CMN Weekly (27 February 2026) - Your Weekly CRISPR...
CMN Weekly
Feb. 27, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

X-Linked Agammaglobulinemia (XLA) is a rare immunodeficiency disorder with a moderate treatment plan, but no cure. This year, Dr David Gray of the Kohn lab at the University of California Los Angeles published a paper inThe CRISPR Journal describing the complex and challenging process of editing the gene responsible, laying the groundwork for pre-clinical studies.
XLA is caused by mutations in the Bruton's tyrosine kinase (BTK) gene. These mutations result in defective B cell receptor (BCR) signalling, which, in turn, impedes the development of B lymphocytes. With no mature B lymphocytes to differentiate into plasma cells to produce immunoglobulin, patients lack antibodies to protect them from pathogens.
XLA sufferers typically endure frequent and recurrent infections, which can be fatal. Treatment options are limited; while patients can receive regular antibody supplementation from healthy donors, there is no long-term treatment option or cure. Allogeneic HSC transplants are occasionally attempted, however, it's usually not worth the risk. With the advent of sophisticated gene-editing technology like CRISPR, gene therapy became a key avenue for XLA research.
Gray cut his teeth working on a gene therapy treatment for sickle cell disease (SCD) in the lab of Professor Donald Kohn. After some time away, he rejoined the Kohn lab in 2015 as a doctoral student, with high hopes of developing a gene-editing treatment for XLA. One of the advantages of working on XLA is that the disease has positive selection – corrected B cells will flourish more than defective ones. This meant that Gray and his colleagues only needed to correct the BTK gene in relatively few cells for those to outcompete the cells that still bear the mutations.
Like SCD, XLA is a monogenic disorder, but that's really where the similarities end. While a single pathogenic mutation causes SCD, XLA can be caused by a myriad of mutations in the BTK gene; for a gene therapy to work for a broad range of patients, the team would have to edit almost the entire gene delivering a functional copy via CRISPR knock-in.
»For XLA, there's a vast range of pathogenic mutations throughout the entire gene; there's not even really a hotspot. So we thought if we're adding in the full-length cDNA at the start of the gene, any patient that has an exonic mutation after that should, in theory, be cured by it,« Gray explains.
As all PhD students know, the road to success is not always that simple. It wasn't long before Gray encountered the first challenges of his PhD project, and he began spending a significant amount of his time in the laboratory troubleshooting and optimising.
»We started in 2015, but [the project] looked a lot different then. The idea was that we're making a cut in the gene, we're adding a functional copy into its normal locus – it should work. But it didn't work out that way at all,« says David Gray.
»So that spawned multiple different aspects of the project. One of them was, how do we improve the amount of integration that we're getting, and the other was how do we improve the expression that we're getting for each integrated copy [of the gene].«
Of course, there were many options available that could have easily increased the expression of BTK, including the addition of exogenous promoters to the donor DNA template for the repair of the gene. However, this wasn't really an option for Gray and his colleagues because evidence suggests that too much BTK in cells can harm patients. Thus, any attempts to increase editing efficiency had to be weighed carefully against the associated risks.
»There's a lot of elements that we can add to this construct to try and crank out [BTK] expression, but this is a disease that already has a moderate treatment plan. So there's room for improvement, but there's a low tolerance for risk. So we had to find this interesting balance where we wanted to improve expression using only endogenous elements as best we could without overshooting,« Gray adds.
B lymphocytes segregate specific antibodies to mark and subsequently destroy pathogens such as influenza viruses. But patients with X-linked Agammaglobulinemia (XLA) don't produce mature B lymphocytes and therefore can't build immunity and are vulnerable to infections. XLA is the first known immune deficiency described in 1952 by Dr Ogden Bruton.
One of the major complicating factors in the project was the sheer size of the BTK gene. Gray used FDA-approved adeno-associated viruses (AAVs) to deliver the CRISPR components needed to repair the gene. As many gene therapy researchers know, AAVs are fantastic vehicles for delivery but are limited by their size – packaging gene-editing components into AAVs is a continual challenge.
The AAVs Gray was using have a 4.7 kilobase maximum size, and the BTK gene is over three kilobases with the homology arms, so there was very little wiggle room. The team found multiple variants that would improve BTK expression, but they wouldn't all fit in the vector, so they began attempting to truncate them.
Another obstacle was finding the right guide RNAs (gRNAs) to perform the edit in the cells of interest successfully. Some gRNAs would work in immortalised cell lines but weren't effective in the peripheral blood stem cells (PBSCs) that Gray needed to deliver the gene to.
»So we started testing new guides directly in primary cells. We still did a lot in cell lines because they're easy to manipulate, but there's always something lost in translation [between immortalised cell lines and primary cells]. We tried dozens of different gRNAs,« Gray says.
Even when Gray managed to integrate the donor DNA template, expression of BTK was still extremely low per integrated copy of the gene.
»That began the most significant troubleshooting part of this project – how do we improve expression without adding exogenous promoters that will drive expression too high,« Gray notes.
An older paper suggested adding terminal introns to boost gene expression. According to Gray, this wasn't a new idea - it's well known that having at least one intron helps with the nuclear export of your mRNA. Interestingly, this paper suggested that adding terminal introns, in particular, was important for transcription termination, nuclear export and polyadenylation.
Unfortunately, the terminal intron of BTK - intron 18 - is massive. Therefore, it would never fit into an AAV with the other necessary gene-editing components. However, it was unclear if the positive effect of adding terminal introns was sequence-based or position-based, and Gray saw an ideal opportunity to test the theory. He decided to add one of the other, smaller introns, into the terminal region of the donor template. This would determine if the positive effect was due to including an intron in the terminal region of the template or because of the sequence of the terminal intron itself.
As Gray discovered, it seems to be the latter – including the entire intron 18 resulted in the best improvement in expression per integrated copy. While adding a smaller intron still helped somewhat, it couldn't boost expression nearly as much as the true terminal intron of the gene. This meant he needed to include intron 18, but space was still an issue. So he decided to truncate intron 18 to see if it would still work. The question was, which parts of the intron really need to be included for it to have a positive effect still?
»There are a few different features of introns known to be important, and we tried to find if there was anything else in intron 18 that could be important. But we didn't find anything. So we decided to take the two ends of the intron, making sure we retained the critical bits, and stick them together to see if it would work,« Gray elucidates.
With this approach, expression increased from 10% to around 40%. While this is not as high as the team had previously achieved with the whole intron 18, it was getting closer to physiological levels of BTK expression, and it seemed like a big step in the right direction.
After testing the terminal intron theory, Gray and the team began a frustrating period of trying everything they could to improve BTK expression whilst still trying to avoid adding exogenous elements.
»At one point, we had to throw in the towel with trying only to use endogenous elements because it just wasn't working,« Gray comments.
After trawling through decades of literature to find something that would help, Gray finally decided to incorporate an exogenous component – woodchuck hepatitis virus post-transcriptional regulatory element (WPRE).
Gray is by no means the first to use WPRE in the gene therapy field, as it's known to stabilise the mRNA message. As he describes, there is still some dispute about the exact mechanism of how WPRE works, but it has been well established in the literature that it does work, and it certainly didn't fail in this case.
»When we added it, we immediately saw a huge increase in expression of BTK. It took us from 10% in the unmodified donor to about 60%. So the next logical step was to try [truncated intron 18 and WPRE] both together. And unsurprisingly, that was the best-expressing donor that we had, getting close to 100% physiological expression,« Gray says.
Again, the solution to one problem came at the sacrifice of creating another; by adding both WPRE and truncated intron 18, the team were again over the AAV vector's packaging limit. The only solution seemed to be to trim intron 18 even further while hoping to retain its 'important bits'.
»I tried going from 600bp in the original truncated version to about 150bp, in what we called the 'micro' version. It still worked – expression took a slight dip, but it was the highest-expressing donor that we were using at that point,« Gray explains.
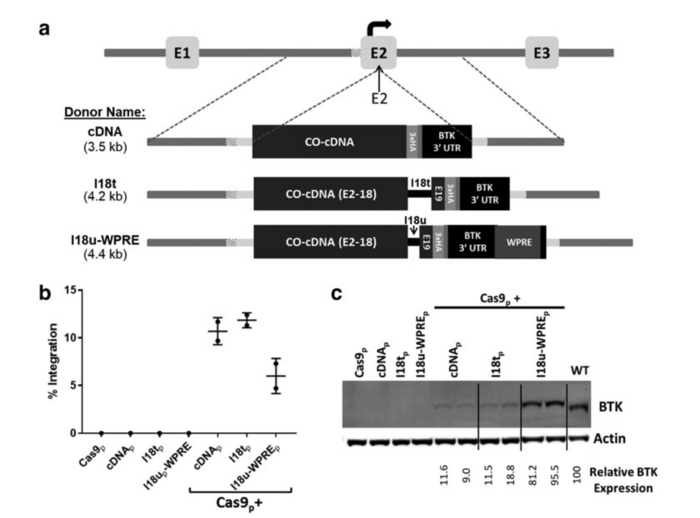
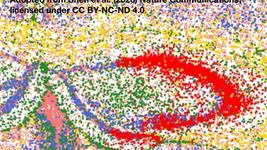
Targeted integration of a corrective cDNA donor into the BTK exon 2 target site led to efficient integration and expression. (a) Schematic of three donor homologous templates designed to integrate into BTK exon 2. (b) BTK-deficient K562 cells were electroporated with the sgRNA/Cas9 plasmid, one of the three BTK exon 2-specific donor templates, or Cas9p together with a donor template. (c) Immunoblot analysis of the treated, BTK-deficient K562 cells probed for actin and the BTK protein. For a detailed description, see Gray et al., The CRISPR Journal (2021).
At this stage of the project, Gray had found the delicate balance between exogenous elements and terminal introns that could provide therapeutically relevant gene integration and expression. But there was one more hurdle to overcome – finding the right gRNAs.
»We discovered that the guides we were using at that point had pretty substantial off-target activity. Something like 40% of the total amount of cuts were off target, which needed to be eliminated. So I was a few years into my thesis, and it was like going back to step one - we had to try a bunch of different guides,« Gray says with a hint of frustration.
While they evaluated the off-target activity of different gRNAs, the team also tried several of the engineered high-fidelity Cas9 nucleases. These worked to slightly different efficiencies with the guides. Still, all of them worked - the worst-performing Cas9 had just 0.5% of activity off target, a significant improvement on the 40% they’d encountered when using a wildtype Cas9.
After years of troubleshooting, optimising, and going back to square one, Gray and his colleagues finally found the perfect combination of elements. They discovered another target site in the gene with a much cleaner profile and a significantly better integration rate in primary cells. Combined with the micro version of intron 18, WPRE, and chemically-modified gRNAs, integration of the corrected BTK was tripled.
The next big challenge is to progress this research into the pre-clinical stage. Gray and his colleagues expect the therapy to be even more successful in human subjects due to positive selection and a more severe disease phenotype. In addition, the use of truncated terminal introns and WPRE is also highly relevant for other studies that aim to edit large genes and increase their expression.
The team haven’t wasted any time – Gray adds that they’ve already been trying to apply these findings to other targets, with some success. Now a postdoctoral researcher at Stonybrook University, Gray works in a different field, but he’s still contributing to ongoing XLA work in the Kohn lab.
»Logically, we hope to see a more profound effect in humans because we have a more severe disease phenotype. The lab is starting to transition into animal studies now and progress into the pre-IND stage. I’m happy to have passed the torch on to such incredible researchers in the Kohn lab,« Gray comments.
Gray hopes his work will not only lead to a cure for XLA but that his hours of troubleshooting and optimising will help smooth the path forward for other students and researchers who are struggling to develop gene therapies.
»I believe this method has promise. Obviously, there’s a lot more testing to be done, but I’m truly hopeful that this will help patients and lead to more cures down the line. On the other side of things, all the troubleshooting we did, I think those findings could help other researchers working on other diseases that have run into the same hurdles. Hopefully, the successes we found after such a long road will help others in the same position.«
Link to original article in The CRISPR Journal:
Rebecca Roberts is a molecular biologist and science writer/communicator based in Queensland, Australia.
ArticleInterviewNewsin vivoAdeno-associated virus (AAV)DiseaseOther Genetic ConditionsRare DiseaseCRISPR-CasCas9