FT819 Remodels Lupus B Cells in Clinical Trial
CMN Briefs
May. 13, 2026


CMN Intelligence - The World’s Most Comprehensive Intelligence Platform for CRISPR-Genomic Medicine and Gene-Editing Clinical Development
Providing market intelligence, data infrastructure, analytics, and reporting services for the global gene-editing sector. Read more...

Around 300 million people around the world are affected by a rare genetic disease. Theoretically, CRISPR can treat most of them by editing the genome to correct the pathogenic variants. However, these 300 million people are split between ~7,000 different diseases that, in some cases, only affect a few handfuls of individuals.
Conventional thinking would lead to the need for 7,000 different CRISPR-based treatments, each targeting a specific genetic variant to cure the affected patients. However, that would not only be a tremendous scientific challenge but would most certainly also lead to a very high price tag for treatments of the rarest of these rare diseases.
At the nonprofit research unit Genethon in Evry just south of Paris, Mario Amendola works hard to solve that problem. He is Group Leader within genome editing and haematopoietic stem cell-based gene therapy, and he has come up with a one-size-fits-all solution.
»Our idea is to find a genetic strategy that can be easily adapted to many different diseases and not only to a single one,« Amendola explains and goes on:
“Our idea is to find a genetic strategy that can be easily adapted to many different diseases and not only to one”Mario Amendola
»There is a group of diseases which are very different from each other, but what they all have in common is the lack of expression of a specific protein in the blood. The specific protein varies from disease to disease, but most of them can be treated in the same way by injecting the missing protein into the blood.«
Current treatments do precisely that, but they come with many limitations, however. So Amendola thought of using genome editing to make blood cells deliver a constant supply of the missing protein. To do that, his team has created an ex vivo CRISPR-based platform for the robust expression of clinically relevant proteins by the erythroid blood-lineage. This is achieved by integrating different transgenes under the control of the endogenous α-globin promoter in human haematopoietic stem/progenitor cells (HSPCs).
»Using this approach, we can plug-in different genes of interest, and we get the same results,« says Amendola and stresses that the strategy can be applied to several diseases.
Mario Amendola and his research team at Genethon delivered proof-of-principle of the strategy in a paper in Nature Communications in July 2020.
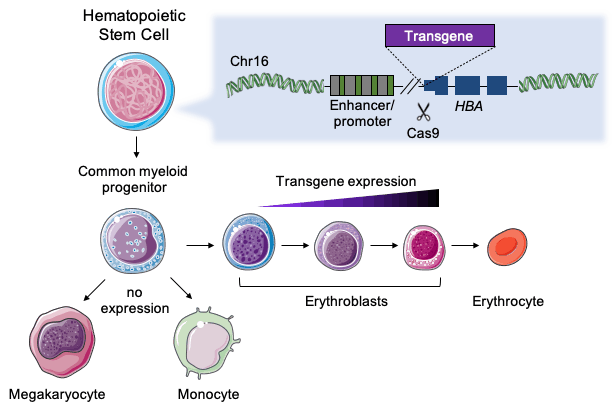
Amendola chose to work with HSPCs since they are a good target for ex vivo editing, which is currently the most studied approach for clinical CRISPR-based therapies. Moreover, when HSPCs have been modified and infused into the patient’s bone marrow, they can produce protein-expressing erythroid cells, i.e., red blood cells, for probably the rest of the patient’s life.
“The α-globin locus is safe because we have four redundant copies of the gene. At the same time, α-globin is constitutively expressed at very high levels only in red blood cells”Mario Amendola
»The α-globin locus is safe because we have four redundant copies of the gene, and clinical data has shown that even if we lose a couple of copies, we don’t have any issues. And at the same time, α-globin is constitutively expressed at very high levels only in red blood cells,« explains the France-based Italian researcher.
The team started by optimising the gene-editing procedure using a promoterless GFP construct and different gRNAs targeting the non-coding sequences of α-globin genes. First, K562 erythroleukaemic cells constitutively expressing Cas9 were edited. Subsequently, select gRNAs were used to transfect human HSPCs with Cas9/gRNA ribonucleoprotein complex (RNP). Choosing a gRNA targeting the 5′UTR region, the team obtained very efficient editing and a ~100-fold GFP upregulation after erythroid induction.
Amendola and his co-workers then tested the optimised platform on four rare genetic disease variants. The absence of functional Factor IX causes haemophilia B, while insufficient levels of α-L-iduronidase, α-galactosidase and lysosomal acid lipase, respectively, cause the three lysosomal storage disorders Hurler syndrome, Fabry disease and Wolman disease. In all four cases, inserting the missing transgenes under control of the endogenous α-globin promoter yielded a 16 to 171-fold increase in mRNA expression, and upon erythroid differentiation, protein levels increased 2.5 to 4.5-fold.
“So the point is not only efficacy, but also safety, and if 20% is OK, there is no reason to go higher”Mario Amendola
To test the modification's therapeutical relevance, conditioned medium from HSPCs edited to express lysosomal acid lipase were exposed to fibroblasts from a patient with Wolman disease. Analyses showed that after three days, the diseased cells had taken up the therapeutic protein, and uptake correlated with decreased cholesterol and lipid deposits, markers of the disease.
Though it is not possible to directly compare these in vitro expression levels with the expression levels in healthy humans, Amendola believes they are within the therapeutic window. He explains:
»We know from previous studies that restoration of 10-20% of wild-type levels is usually enough to be healthy, and on the other hand, 2 to 3-fold overexpression is often OK. So the point is not only efficacy, but also safety, and if 20% is OK, there is no reason to go much higher.«
To test the long-term function of edited cells in vivo, Amendola and his team transplanted HSPCs edited to express lysosomal acid lipase in mice. The results showed successful engraftment and repopulation in bone marrow, spleen and blood. In addition, the HSPCs could give rise to progenitors and differentiated into haematopoietic lineages.
Last Friday, the researchers at Genethon showed in another paper in Blood Advances that a similar approach can be adapted to treat a group of blood disorders, β-thalassemia, caused by mutations in the β-globin gene cluster.
α-and β-globins normally associate to form adult haemoglobin, but when expression of the two proteins is not balanced, α-globins precipitate and damage cell membranes, causing haemolysis and ineffective erythropoiesis.
Therefore, the researchers decided to combine the gene knock-in approach with gene knockout in order to balance the expression of two essential proteins.
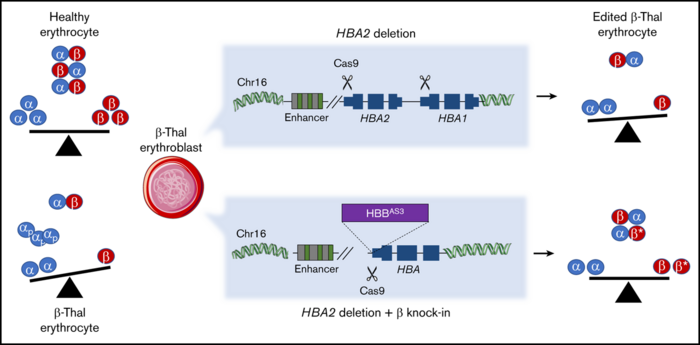
The researchers inserted a β-globin transgene behind the endogenous α-globin promoter. That effectively killed two birds with one stone: α-globins were downregulated, while β-globins were upregulated. This approach might restore the balance of the two proteins in β-thalassemia patients where α-globins are in excess. The strategy was tested in HSPCs from patients and resulted in the correction of the α/β globin imbalance in the subsequently derived erythroblasts.
Mario Amendola is optimistic that his common gene-editing platform for treating several rare genetic diseases will be suitable for future clinical use. While optimising the gene-editing protocol, the team used puromycin resistance for selection and GFP as a marker, but that was only for experimental use. In the final protocol, no selection or marker genes are necessary, and only the therapeutic transgene will be integrated into the patients HSPCs. Amendola explains:
»Our gene-editing protocol is very efficient, so we can easily edit more than 50% of the HSPCs ex vivo without any selection. In most cases, our goal is to modify as little as possible and just reach the therapeutic window. We know we can go very high, but the final goal is to go very low.«
“We know we can go very high, but the final goal is to go very low”Mario Amendola
Before moving into clinical trials, however, more preclinical in vivo experiments in animals are needed. Also, the team needs to select a single disease to work with, and that will probably be lysosomal storage disorder, Amendola says. Moreover, while CRISPR in HSPCs is used in ongoing clinical trials, this has so far not been the case in combination with targeted transgene integration.
»This means we will need to provide even more data to validate the technique for the first time. And we also need to get more data on the disease,« says Amendola.
Link to the original article in Nature Communications from July 2020:
Ex vivo editing of human hematopoietic stem cells for erythroid expression of therapeutic proteins
Link to the original article in Blood Advances from February 2021:
ArticleInterviewNewsBeta ThalassemiaHaemophilia BRare DiseaseCRISPR-CasCas9Généthon





